表达数量性状位点(expression quantitative trait loci,eQTL)是指基因组中的特定位点,这些位点上的遗传变异能够影响基因表达水平的变异。eQTL研究是基因组学研究中的一个重要领域,它有助于我们理解基因表达的遗传基础以及复杂性状的分子机制。
eQTL可以分为两大类:
- 顺式eQTL (cis-eQTL):这类eQTL位于基因表达变化所在的染色体区域内,通常接近或在基因的附近(比如1Mb范围内)。顺式作用eQTL可能涉及基因的启动子或增强子区域的变异,影响基因的转录效率。
- 反式eQTL (trans-eQTL):与顺式eQTL不同,反式eQTL位于距离基因较远的染色体区域,甚至可能位于不同的染色体上。反式作用eQTL可能通过远程调控元件,如远程增强子或转录因子,来影响基因表达。
eQTL分析的一般流程包括:
- 数据收集:需要样本的基因表达数据、基因型数据以及可能的样本表型信息。
- 数据预处理:包括数据的质量控制、标准化等步骤。
- 统计分析:通常使用线性回归模型来识别与基因表达水平显著相关的SNP位点。模型可能会包括其他协变量,如性别、年龄或批次效应,以校正混杂因素。
- 结果解释:识别出显著的eQTL后,进一步分析其生物学意义和可能的分子机制。
进行eQTL分析的R包包括但不限于:
- MatrixEQTL:一个用于快速进行大规模eQTL分析的R包,支持多种遗传模型和错误结构。 eQTpLot:一个用户友好的R包,用于eQTL和GWAS信号的可视化和共定位。
eQTL研究的结果可以帮助我们理解遗传变异如何影响基因表达,进而影响个体的性状和疾病风险。此外,eQTL数据还可以用于基因功能的研究、疾病机制的探索以及药物靶点的发现。
值得注意的是,eQTL效应可能具有人群、组织和细胞类型的特异性,这意味着不同背景下eQTL效应可能不同。因此,在应用eQTL结果时需要考虑这些因素。
全基因组关联研究(Genome-Wide Association Study,GWAS)是一种在全基因组范围内识别与特定表型或疾病相关联的遗传变异的研究方法。GWAS通过检测大量个体的遗传变异(通常是单核苷酸多态性,或称为SNPs),来找出与疾病或性状相关的遗传标记。
GWAS的主要步骤包括:
样本收集:收集足够数量的个体样本,包括患病个体和对照个体。
基因分型:利用高密度的SNP芯片对个体的全基因组进行分型,以获取遗传信息。
质量控制:对基因分型数据进行质量控制,包括去除低质量的SNP位点和样本,校正人群分层等。
统计分析:使用统计方法(如线性回归、逻辑回归等)对每个SNP位点与表型之间的关联进行检验。
结果解释:对统计显著的SNP位点进行解释,以识别可能的疾病相关基因或遗传区域。
验证研究:在独立的样本中对显著的关联结果进行验证,以确认其可靠性。
生物信息学分析:对GWAS结果进行更深入的生物信息学分析,如路径分析、网络分析等,以理解遗传变异如何影响疾病机制。
GWAS的应用: GWAS已被广泛应用于多种复杂疾病的遗传研究,包括但不限于阿尔茨海默病、乳腺癌、糖尿病、冠心病、肺癌、前列腺癌和肥胖等。通过GWAS,科学家们已经鉴定出与这些疾病相关的多个易感基因。
GWAS的挑战和限制: 样本量:GWAS需要非常大的样本量来提高统计功效,确定可复现的全基因组显著关联。 混杂因素:如人群分层、年龄、性别等混杂因素可能影响GWAS的结果。 多重检验:由于测试大量的SNP位点,需要进行多重检验校正,这可能导致错过一些真实的关联信号。 罕见变异:GWAS主要检测常见的遗传变异,对罕见变异的检测能力有限。
Code
# install.packages("MatrixEQTL")
library(MatrixEQTL)
path <- find.package("MatrixEQTL")
path
#> [1] "D:/R-4.3.3/library/MatrixEQTL"
# 把"MatrixEQTL"包的data复制到当前工作目录
expr <- read.table("data/GE.txt",sep = "\t",header = T,row.names = 1)
expr
| Gene_01 |
4.91 |
4.63 |
5.18 |
5.07 |
5.74 |
5.09 |
5.31 |
5.29 |
4.73 |
5.72 |
4.75 |
4.54 |
5.01 |
5.03 |
4.84 |
4.44 |
| Gene_02 |
13.78 |
13.14 |
13.18 |
13.04 |
12.85 |
13.07 |
13.09 |
12.83 |
14.94 |
13.86 |
15.26 |
14.73 |
14.08 |
14.33 |
14.72 |
14.97 |
| Gene_03 |
12.06 |
12.29 |
13.07 |
13.65 |
13.86 |
12.84 |
12.29 |
13.03 |
13.13 |
14.93 |
12.40 |
13.38 |
13.70 |
14.49 |
14.14 |
13.35 |
| Gene_04 |
11.63 |
11.88 |
12.74 |
12.66 |
13.16 |
11.99 |
11.97 |
12.81 |
11.74 |
13.29 |
10.88 |
11.37 |
12.09 |
12.50 |
11.40 |
11.45 |
| Gene_05 |
14.72 |
14.66 |
14.63 |
15.91 |
15.46 |
14.74 |
15.17 |
15.01 |
14.05 |
14.90 |
12.96 |
13.56 |
14.06 |
14.44 |
14.12 |
13.76 |
| Gene_06 |
12.29 |
12.23 |
12.47 |
13.21 |
12.63 |
12.18 |
12.44 |
12.45 |
13.22 |
12.70 |
12.93 |
12.84 |
13.20 |
13.19 |
12.64 |
12.80 |
| Gene_07 |
12.56 |
12.71 |
12.49 |
13.41 |
13.60 |
12.35 |
12.27 |
13.42 |
14.73 |
15.47 |
13.85 |
14.31 |
15.25 |
15.55 |
14.90 |
14.04 |
| Gene_08 |
12.27 |
12.58 |
12.61 |
13.02 |
12.86 |
12.32 |
12.30 |
13.19 |
15.21 |
14.60 |
14.72 |
14.56 |
14.98 |
14.92 |
14.53 |
14.72 |
| Gene_09 |
9.82 |
9.29 |
8.95 |
8.18 |
8.11 |
9.43 |
9.17 |
9.25 |
10.95 |
9.82 |
12.60 |
11.99 |
11.20 |
10.31 |
11.13 |
11.69 |
| Gene_10 |
14.24 |
14.52 |
14.62 |
13.65 |
13.46 |
14.04 |
13.97 |
13.73 |
12.29 |
12.01 |
13.47 |
13.30 |
12.34 |
12.23 |
12.80 |
12.47 |
Code
snp <- read.table("data/SNP.txt",sep = "\t",header = T,row.names = 1)
snp
| Snp_01 |
2 |
0 |
2 |
0 |
2 |
1 |
2 |
1 |
1 |
1 |
2 |
2 |
1 |
2 |
2 |
1 |
| Snp_02 |
0 |
1 |
1 |
2 |
2 |
1 |
0 |
0 |
0 |
1 |
1 |
1 |
1 |
0 |
1 |
1 |
| Snp_03 |
1 |
0 |
1 |
0 |
1 |
1 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
1 |
1 |
2 |
| Snp_04 |
0 |
1 |
2 |
2 |
2 |
1 |
1 |
0 |
0 |
0 |
1 |
2 |
1 |
1 |
1 |
0 |
| Snp_05 |
1 |
1 |
2 |
1 |
1 |
2 |
1 |
1 |
0 |
1 |
1 |
2 |
0 |
1 |
2 |
1 |
| Snp_06 |
2 |
2 |
2 |
1 |
1 |
0 |
1 |
0 |
2 |
1 |
1 |
1 |
2 |
0 |
2 |
1 |
| Snp_07 |
1 |
1 |
2 |
2 |
0 |
1 |
1 |
1 |
1 |
0 |
2 |
2 |
0 |
1 |
1 |
1 |
| Snp_08 |
1 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
1 |
1 |
0 |
2 |
0 |
1 |
1 |
1 |
| Snp_09 |
2 |
1 |
2 |
2 |
0 |
1 |
1 |
0 |
2 |
1 |
1 |
0 |
1 |
1 |
0 |
0 |
| Snp_10 |
1 |
1 |
0 |
0 |
0 |
2 |
2 |
1 |
1 |
2 |
1 |
1 |
1 |
1 |
1 |
0 |
| Snp_11 |
2 |
2 |
2 |
0 |
2 |
1 |
1 |
2 |
1 |
2 |
0 |
1 |
0 |
1 |
1 |
2 |
| Snp_12 |
1 |
1 |
2 |
2 |
2 |
1 |
1 |
1 |
1 |
0 |
2 |
0 |
1 |
1 |
0 |
2 |
| Snp_13 |
0 |
1 |
1 |
1 |
1 |
1 |
2 |
1 |
2 |
2 |
0 |
0 |
0 |
1 |
1 |
1 |
| Snp_14 |
0 |
0 |
1 |
0 |
1 |
2 |
2 |
2 |
1 |
1 |
1 |
0 |
1 |
0 |
1 |
0 |
| Snp_15 |
1 |
2 |
1 |
2 |
2 |
1 |
2 |
1 |
1 |
2 |
1 |
1 |
1 |
2 |
0 |
2 |
Code
covar <- read.table("data/Covariates.txt",sep = "\t",header = T,row.names = 1)
covar
| gender |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
| age |
36 |
40 |
46 |
65 |
69 |
43 |
40 |
54 |
44 |
70 |
24 |
34 |
55 |
62 |
48 |
40 |
Code
e1 <- as.numeric(expr[1,])
s1 <- as.numeric(snp[1,])
e1
#> [1] 4.91 4.63 5.18 5.07 5.74 5.09 5.31 5.29 4.73 5.72 4.75 4.54 5.01 5.03 4.84
#> [16] 4.44
s1
#> [1] 2 0 2 0 2 1 2 1 1 1 2 2 1 2 2 1
lm1 <- lm(e1~s1)
broom::tidy(lm1)
| (Intercept) |
4.929677 |
0.2137508 |
23.0627348 |
0.0000000 |
| s1 |
0.063871 |
0.1386998 |
0.4604979 |
0.6522304 |
Code
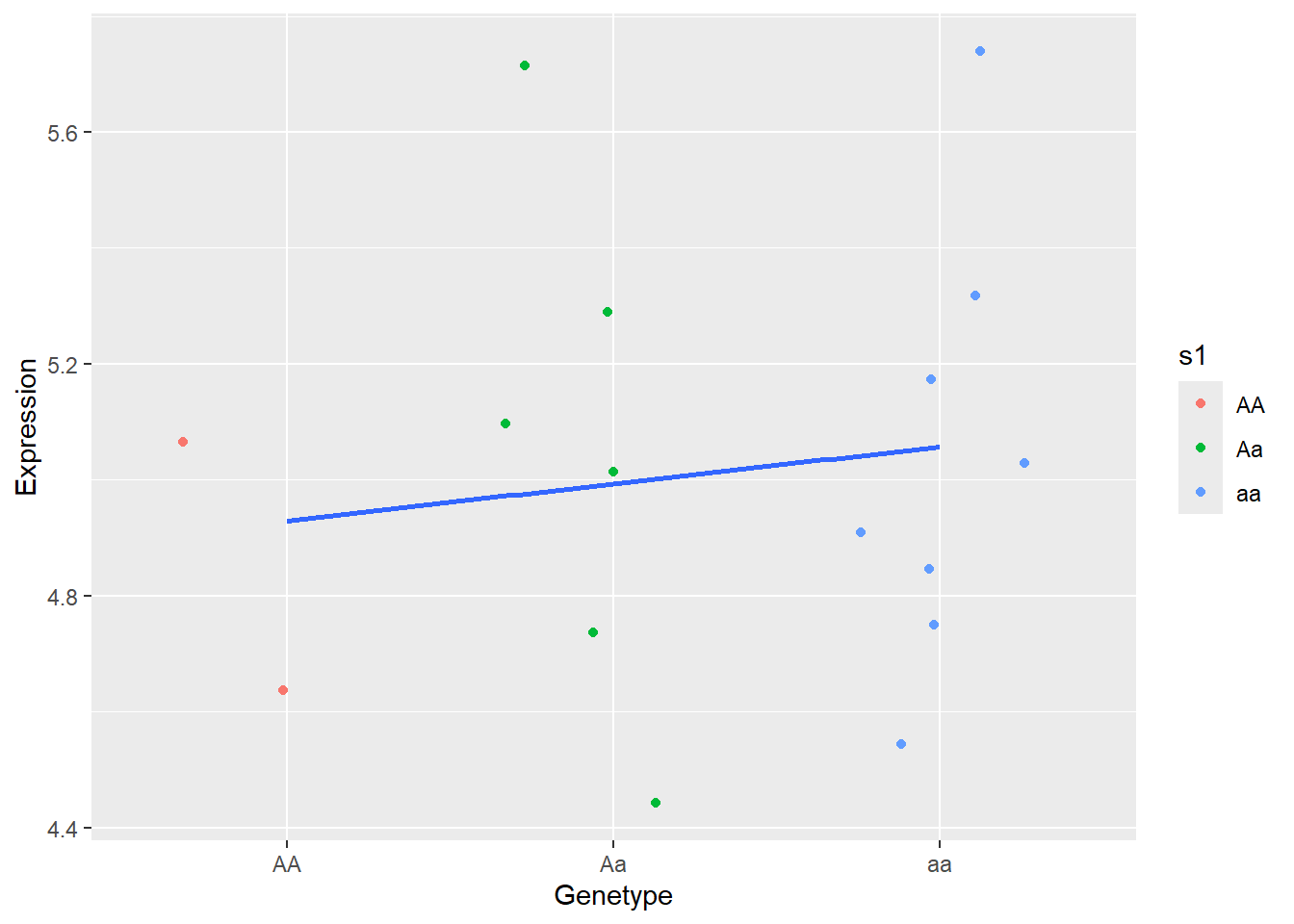
ggplot(data = tibble(e1,
s1=factor(s1,labels = c("AA","Aa","aa"))
),
mapping = aes(x=s1,y=e1)
)+
geom_point(aes(color=s1),position = position_jitter())+
geom_smooth(aes(group=1),formula = "y~x",method = "lm",se=F)+
xlab("Genetype")+
ylab("Expression")
Code
# 设置参数
pOutputThreshold <- 1e-2
errorCovariance <- numeric()
useModel <- modelLINEAR
### SNP
snp <- SlicedData$new()
snp$fileDelimiter <- "\t"
snp$fileOmitCharacters <- "NA"
snp$fileSkipRows <- 1 # 跳过列名
snp$fileSkipColumns <- 1 # 跳过行名
snp$fileSliceSize <- 2000 # nrows
snp$LoadFile("data/SNP.txt")
snp
#> SlicedData object. For more information type: ?SlicedData
#> Number of columns: 16
#> Number of rows: 15
#> Data is stored in 1 slices
#> Top left corner of the first slice (up to 10x10):
#> Sam_01 Sam_02 Sam_03 Sam_04 Sam_05 Sam_06 Sam_07 Sam_08 Sam_09 Sam_10
#> Snp_01 2 0 2 0 2 1 2 1 1 1
#> Snp_02 0 1 1 2 2 1 0 0 0 1
#> Snp_03 1 0 1 0 1 1 1 1 0 1
#> Snp_04 0 1 2 2 2 1 1 0 0 0
#> Snp_05 1 1 2 1 1 2 1 1 0 1
#> Snp_06 2 2 2 1 1 0 1 0 2 1
#> Snp_07 1 1 2 2 0 1 1 1 1 0
#> Snp_08 1 0 1 0 1 0 0 1 1 1
#> Snp_09 2 1 2 2 0 1 1 0 2 1
#> Snp_10 1 1 0 0 0 2 2 1 1 2
### gene
gene <- SlicedData$new()
gene$fileDelimiter <- "\t"
gene$fileOmitCharacters <- "NA"
gene$fileSkipRows <- 1
gene$fileSkipColumns <- 1
gene$fileSliceSize <- 2000
gene$LoadFile("data/GE.txt")
gene
#> SlicedData object. For more information type: ?SlicedData
#> Number of columns: 16
#> Number of rows: 10
#> Data is stored in 1 slices
#> Top left corner of the first slice (up to 10x10):
#> Sam_01 Sam_02 Sam_03 Sam_04 Sam_05 Sam_06 Sam_07 Sam_08 Sam_09 Sam_10
#> Gene_01 4.91 4.63 5.18 5.07 5.74 5.09 5.31 5.29 4.73 5.72
#> Gene_02 13.78 13.14 13.18 13.04 12.85 13.07 13.09 12.83 14.94 13.86
#> Gene_03 12.06 12.29 13.07 13.65 13.86 12.84 12.29 13.03 13.13 14.93
#> Gene_04 11.63 11.88 12.74 12.66 13.16 11.99 11.97 12.81 11.74 13.29
#> Gene_05 14.72 14.66 14.63 15.91 15.46 14.74 15.17 15.01 14.05 14.90
#> Gene_06 12.29 12.23 12.47 13.21 12.63 12.18 12.44 12.45 13.22 12.70
#> Gene_07 12.56 12.71 12.49 13.41 13.60 12.35 12.27 13.42 14.73 15.47
#> Gene_08 12.27 12.58 12.61 13.02 12.86 12.32 12.30 13.19 15.21 14.60
#> Gene_09 9.82 9.29 8.95 8.18 8.11 9.43 9.17 9.25 10.95 9.82
#> Gene_10 14.24 14.52 14.62 13.65 13.46 14.04 13.97 13.73 12.29 12.01
# 协变量
covar <- SlicedData$new()
covar$fileDelimiter <- "\t"
covar$fileOmitCharacters <- "NA"
covar$fileSkipRows <- 1
covar$fileSkipColumns <- 1
covar$fileSliceSize <- 2000
covar$LoadFile("data/Covariates.txt")
covar
#> SlicedData object. For more information type: ?SlicedData
#> Number of columns: 16
#> Number of rows: 2
#> Data is stored in 1 slices
#> Top left corner of the first slice (up to 10x10):
#> Sam_01 Sam_02 Sam_03 Sam_04 Sam_05 Sam_06 Sam_07 Sam_08 Sam_09 Sam_10
#> gender 0 0 0 0 0 0 0 0 1 1
#> age 36 40 46 65 69 43 40 54 44 70
Code
eqtl <- Matrix_eQTL_engine(
snps = snp,
gene = gene,
cvrt = covar,
output_file_name = "",
pvOutputThreshold = pOutputThreshold,
useModel = useModel,
errorCovariance = errorCovariance,
verbose = T,
pvalue.hist = T,
min.pv.by.genesnp = F,
noFDRsaveMemory = F
)
Code
eqtl$all
#> $ntests
#> [1] 150
#>
#> $neqtls
#> [1] 5
#>
#> $eqtls
#> snps gene statistic pvalue FDR beta
#> 1 Snp_05 Gene_03 38.812160 5.515519e-14 8.273279e-12 0.4101317
#> 2 Snp_13 Gene_09 -3.914403 2.055817e-03 1.541863e-01 -0.2978847
#> 3 Snp_11 Gene_06 -3.221962 7.327756e-03 2.853368e-01 -0.2332470
#> 4 Snp_04 Gene_10 3.201666 7.608981e-03 2.853368e-01 0.2321123
#> 5 Snp_14 Gene_01 3.070005 9.716705e-03 2.915011e-01 0.2147077
#>
#> $hist.bins
#> [1] 0.00 0.01 0.02 0.03 0.04 0.05 0.06 0.07 0.08 0.09 0.10 0.11 0.12 0.13 0.14
#> [16] 0.15 0.16 0.17 0.18 0.19 0.20 0.21 0.22 0.23 0.24 0.25 0.26 0.27 0.28 0.29
#> [31] 0.30 0.31 0.32 0.33 0.34 0.35 0.36 0.37 0.38 0.39 0.40 0.41 0.42 0.43 0.44
#> [46] 0.45 0.46 0.47 0.48 0.49 0.50 0.51 0.52 0.53 0.54 0.55 0.56 0.57 0.58 0.59
#> [61] 0.60 0.61 0.62 0.63 0.64 0.65 0.66 0.67 0.68 0.69 0.70 0.71 0.72 0.73 0.74
#> [76] 0.75 0.76 0.77 0.78 0.79 0.80 0.81 0.82 0.83 0.84 0.85 0.86 0.87 0.88 0.89
#> [91] 0.90 0.91 0.92 0.93 0.94 0.95 0.96 0.97 0.98 0.99 1.00
#>
#> $hist.counts
#> [1] 5 0 3 1 0 1 3 1 1 5 4 2 0 1 2 1 1 1 0 0 1 0 2 1 4 1 2 1 2 0 2 3 2 0 2 2 4
#> [38] 1 1 1 0 0 0 2 1 1 3 1 0 1 1 3 0 1 2 2 1 0 2 0 2 2 2 2 0 1 2 2 3 2 2 0 2 2
#> [75] 1 3 2 2 0 1 1 1 2 1 5 1 3 0 2 1 2 1 1 2 2 2 1 1 2 1
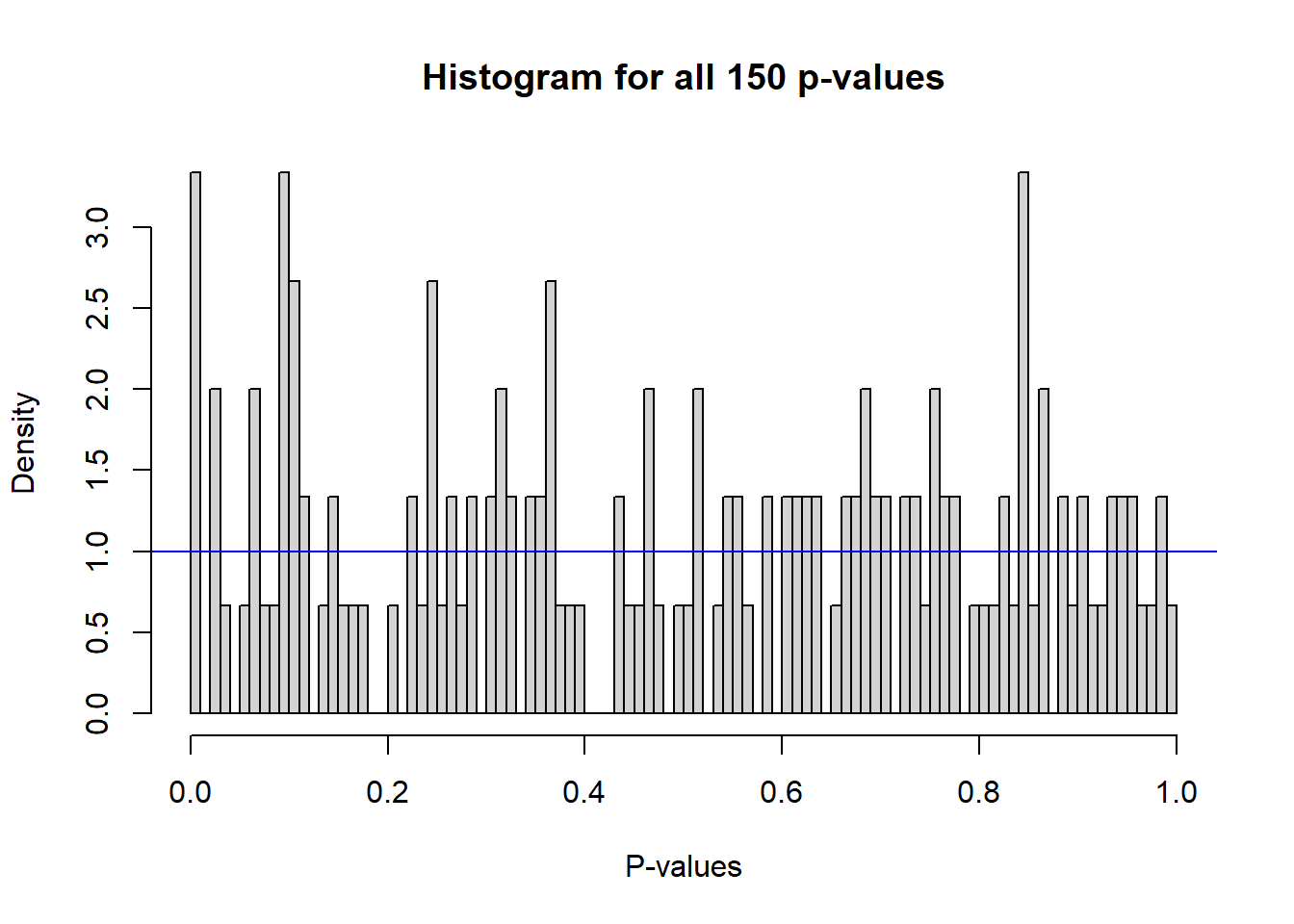
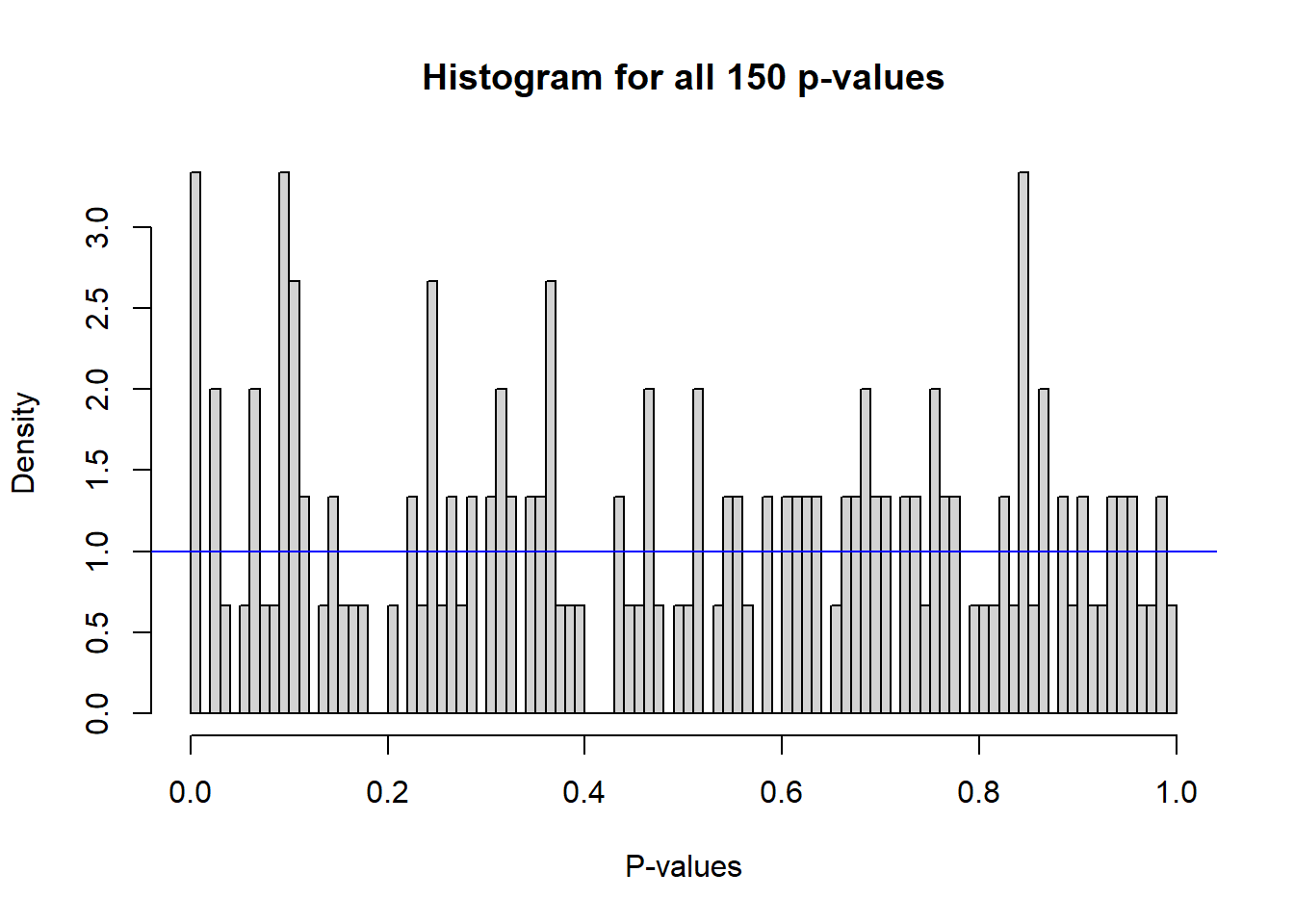
plot(eqtl)
Code
#===================Results==================================#
cat('Analysis done in: ', eqtl$time.in.sec, ' seconds', '\n');
#> Analysis done in: 0.02 seconds
cat('Detected eQTLs:', '\n');
#> Detected eQTLs:
show(eqtl$all$eqtls)
#> snps gene statistic pvalue FDR beta
#> 1 Snp_05 Gene_03 38.812160 5.515519e-14 8.273279e-12 0.4101317
#> 2 Snp_13 Gene_09 -3.914403 2.055817e-03 1.541863e-01 -0.2978847
#> 3 Snp_11 Gene_06 -3.221962 7.327756e-03 2.853368e-01 -0.2332470
#> 4 Snp_04 Gene_10 3.201666 7.608981e-03 2.853368e-01 0.2321123
#> 5 Snp_14 Gene_01 3.070005 9.716705e-03 2.915011e-01 0.2147077
## Plot the histogram of all p-values
plot(eqtl)
Bioinformatics Bioinformatics 17 Biostrings包 参考资料 上游分析 1 Sequences Data to Expression Matrix 单细胞分析 2 写在前面 3 获取scRNA-seq表达值矩阵 4 SingleCellExperiment 5 工作流 6 质量控制 7 标准化 8 特征选择 9 降维 10 聚类 11 conflicts_prefer(dplyr::filter) 12 细胞类型注释 Seurat 13 Seurat 下游分析 14 差异表达基因分析 15 富集分析 16 伪时序分析 基因组学 17 Biostrings包 18 eQTL 基因组学 18 eQTL
```{r include=FALSE}
conflicts_prefer(GenomicRanges::setdiff)
```
# eQTL
**表达数量性状位点(expression quantitative trait loci,eQTL)**是指基因组中的特定位点,这些位点上的遗传变异能够影响基因表达水平的变异。eQTL研究是基因组学研究中的一个重要领域,它有助于我们理解基因表达的遗传基础以及复杂性状的分子机制。
eQTL可以分为两大类:
- 顺式eQTL (cis-eQTL):这类eQTL位于基因表达变化所在的染色体区域内,通常接近或在基因的附近(比如1Mb范围内)。顺式作用eQTL可能涉及基因的启动子或增强子区域的变异,影响基因的转录效率。
- 反式eQTL (trans-eQTL):与顺式eQTL不同,反式eQTL位于距离基因较远的染色体区域,甚至可能位于不同的染色体上。反式作用eQTL可能通过远程调控元件,如远程增强子或转录因子,来影响基因表达。
eQTL分析的一般流程包括:
1. 数据收集:需要样本的基因表达数据、基因型数据以及可能的样本表型信息。
2. 数据预处理:包括数据的质量控制、标准化等步骤。
3. 统计分析:通常使用线性回归模型来识别与基因表达水平显著相关的SNP位点。模型可能会包括其他协变量,如性别、年龄或批次效应,以校正混杂因素。
4. 结果解释:识别出显著的eQTL后,进一步分析其生物学意义和可能的分子机制。
进行eQTL分析的R包包括但不限于:
- MatrixEQTL:一个用于快速进行大规模eQTL分析的R包,支持多种遗传模型和错误结构。 eQTpLot:一个用户友好的R包,用于eQTL和GWAS信号的可视化和共定位。
eQTL研究的结果可以帮助我们理解遗传变异如何影响基因表达,进而影响个体的性状和疾病风险。此外,eQTL数据还可以用于基因功能的研究、疾病机制的探索以及药物靶点的发现。
值得注意的是,eQTL效应可能具有人群、组织和细胞类型的特异性,这意味着不同背景下eQTL效应可能不同。因此,在应用eQTL结果时需要考虑这些因素。
**全基因组关联研究(Genome-Wide Association Study,GWAS)**是一种在全基因组范围内识别与特定表型或疾病相关联的遗传变异的研究方法。GWAS通过检测大量个体的遗传变异(通常是单核苷酸多态性,或称为SNPs),来找出与疾病或性状相关的遗传标记。
GWAS的主要步骤包括:
1. 样本收集:收集足够数量的个体样本,包括患病个体和对照个体。
2. 基因分型:利用高密度的SNP芯片对个体的全基因组进行分型,以获取遗传信息。
3. 质量控制:对基因分型数据进行质量控制,包括去除低质量的SNP位点和样本,校正人群分层等。
4. 统计分析:使用统计方法(如线性回归、逻辑回归等)对每个SNP位点与表型之间的关联进行检验。
5. 结果解释:对统计显著的SNP位点进行解释,以识别可能的疾病相关基因或遗传区域。
6. 验证研究:在独立的样本中对显著的关联结果进行验证,以确认其可靠性。
7. 生物信息学分析:对GWAS结果进行更深入的生物信息学分析,如路径分析、网络分析等,以理解遗传变异如何影响疾病机制。
GWAS的应用: GWAS已被广泛应用于多种复杂疾病的遗传研究,包括但不限于阿尔茨海默病、乳腺癌、糖尿病、冠心病、肺癌、前列腺癌和肥胖等。通过GWAS,科学家们已经鉴定出与这些疾病相关的多个易感基因。
GWAS的挑战和限制: 样本量:GWAS需要非常大的样本量来提高统计功效,确定可复现的全基因组显著关联。 混杂因素:如人群分层、年龄、性别等混杂因素可能影响GWAS的结果。 多重检验:由于测试大量的SNP位点,需要进行多重检验校正,这可能导致错过一些真实的关联信号。 罕见变异:GWAS主要检测常见的遗传变异,对罕见变异的检测能力有限。
```{r snpdata}
# install.packages("MatrixEQTL")
library(MatrixEQTL)
path <- find.package("MatrixEQTL")
path
# 把"MatrixEQTL"包的data复制到当前工作目录
expr <- read.table("data/GE.txt",sep = "\t",header = T,row.names = 1)
expr
snp <- read.table("data/SNP.txt",sep = "\t",header = T,row.names = 1)
snp
covar <- read.table("data/Covariates.txt",sep = "\t",header = T,row.names = 1)
covar
```
quarto-executable-code-5450563D
```r
e1 <- as.numeric(expr[1,])
s1 <- as.numeric(snp[1,])
e1
s1
lm1 <- lm(e1~s1)
broom::tidy(lm1)
ggplot(data = tibble(e1,
s1=factor(s1,labels = c("AA","Aa","aa"))
),
mapping = aes(x=s1,y=e1)
)+
geom_point(aes(color=s1),position = position_jitter())+
geom_smooth(aes(group=1),formula = "y~x",method = "lm",se=F)+
xlab("Genetype")+
ylab("Expression")
```
quarto-executable-code-5450563D
```r
# 设置参数
pOutputThreshold <- 1e-2
errorCovariance <- numeric()
useModel <- modelLINEAR
### SNP
snp <- SlicedData$new()
snp$fileDelimiter <- "\t"
snp$fileOmitCharacters <- "NA"
snp$fileSkipRows <- 1 # 跳过列名
snp$fileSkipColumns <- 1 # 跳过行名
snp$fileSliceSize <- 2000 # nrows
snp$LoadFile("data/SNP.txt")
snp
### gene
gene <- SlicedData$new()
gene$fileDelimiter <- "\t"
gene$fileOmitCharacters <- "NA"
gene$fileSkipRows <- 1
gene$fileSkipColumns <- 1
gene$fileSliceSize <- 2000
gene$LoadFile("data/GE.txt")
gene
# 协变量
covar <- SlicedData$new()
covar$fileDelimiter <- "\t"
covar$fileOmitCharacters <- "NA"
covar$fileSkipRows <- 1
covar$fileSkipColumns <- 1
covar$fileSliceSize <- 2000
covar$LoadFile("data/Covariates.txt")
covar
```
```{r eQTL}
eqtl <- Matrix_eQTL_engine(
snps = snp,
gene = gene,
cvrt = covar,
output_file_name = "",
pvOutputThreshold = pOutputThreshold,
useModel = useModel,
errorCovariance = errorCovariance,
verbose = T,
pvalue.hist = T,
min.pv.by.genesnp = F,
noFDRsaveMemory = F
)
```
quarto-executable-code-5450563D
```r
eqtl$all
plot(eqtl)
```
quarto-executable-code-5450563D
```r
#===================Results==================================#
cat('Analysis done in: ', eqtl$time.in.sec, ' seconds', '\n');
cat('Detected eQTLs:', '\n');
show(eqtl$all$eqtls)
## Plot the histogram of all p-values
plot(eqtl)
```