Code
library(arrow, warn.conflicts = FALSE)
logCount <- read_tsv_arrow("data/DEGs_Analysis/TCGA-LUAD.htseq_counts.tsv.gz")
# 读取计数矩阵 log2(count+1) # 60488 个gene × 586 个samplehttps://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html
肺腺癌
Lung Adenocarcinoma (LUAD) gene expression RNAseq HTSeq - Counts (n=585) GDC 中心 和Phenotype (n=877)
library(arrow, warn.conflicts = FALSE)
logCount <- read_tsv_arrow("data/DEGs_Analysis/TCGA-LUAD.htseq_counts.tsv.gz")
# 读取计数矩阵 log2(count+1) # 60488 个gene × 586 个sample如果分析总的转录本,不需要把mRNA、lncRNA等挑出来,所以下载的这个自带注释文件ID/Gene Mapping
gtf_v22_transcripts <- read_delim("data/DEGs_Analysis/gencode.v22.annotation.gene.probeMap")如果只想用蛋白质编码(protein_coding)相关基因,用全基因编码注释文件 gencode.v46.annotation.gtf.gz
# 全基因编码 BiocManager::install("rtracklayer")
gtf_v46 <- rtracklayer::import('data/DEGs_Analysis/gencode.v46.annotation.gtf.gz') |>
as_tibble()
distinct(gtf_v46,gene_type)
distinct(gtf_v46,gene_name)
# 选择 蛋白编码mRNA相关基因
gtf_v46_mRNA <- dplyr::select(gtf_v46,
c("gene_id","gene_type", "gene_name"))|>
dplyr::filter(gene_type == "protein_coding") |>
distinct()
head(gtf_v46_mRNA )
write.csv(gtf_v46_mRNA,"data/gtf_v46_mRNA.csv")gene_logCount <- logCount |>
inner_join(gtf_v22_transcripts, by = c("Ensembl_ID" = "id")) |>
dplyr::select(gene, starts_with("TCGA") )dim(gene_logCount)
#> [1] 60483 586
gene_logCount[1:6,1:3]| gene | TCGA-97-7938-01A | TCGA-55-7574-01A |
|---|---|---|
| TSPAN6 | 10.989394 | 9.967226 |
| TNMD | 4.000000 | 0.000000 |
| DPM1 | 10.253847 | 9.541097 |
| SCYL3 | 9.776433 | 9.131857 |
| C1orf112 | 7.971544 | 8.348728 |
| FGR | 8.724514 | 9.703904 |
先去对数 log2(count+1)
基因名是有重复的,查看重复情况
根据基因名分组,重复则取平均值去重,去掉重复的行
library(data.table)
setDT(gene_Count)
Counts <- gene_Count[,lapply(.SD,mean),by=gene, .SDcols=patterns("^(TCGA)")]Counts <- Counts |> column_to_rownames("gene")
dim(Counts)
#> [1] 58387 585| g | x | y |
|---|---|---|
| A | 1 | 3 |
| B | 2 | 4 |
| A | 3 | 5 |
| C | 4 | 6 |
| C | 5 | 7 |
# 数据多太慢了
df|> group_by(g) |> summarise_all(list(mean))| g | x | y |
|---|---|---|
| A | 2.0 | 4.0 |
| B | 2.0 | 4.0 |
| C | 4.5 | 6.5 |
| g | x | y |
|---|---|---|
| A | 2.0 | 4.0 |
| B | 2.0 | 4.0 |
| C | 4.5 | 6.5 |
提取lncRNA、miRNA等也是类似的操作
#首先载入下载好胆管癌的样本信息文件
# 877 个sample × 125 个样本变量
colData <- fread("data/DEGs_Analysis/TCGA-LUAD.GDC_phenotype.tsv.gz", sep = "\t", header = TRUE)
# 选择样本 放疗与kras基因
table(colData$radiation_therapy,colData$kras_mutation_found)
#>
#> NO YES
#> 230 1 3
#> NO 491 41 24
#> YES 72 11 4
pf <- colData |> dplyr::filter(kras_mutation_found!= "") |>
dplyr::filter(radiation_therapy!= "") |>
dplyr::select(submitter_id.samples,submitter_id,radiation_therapy,kras_mutation_found)
xtabs(formula = ~radiation_therapy+kras_mutation_found,data = pf,subset = NULL)
#> kras_mutation_found
#> radiation_therapy NO YES
#> NO 41 24
#> YES 11 4到这里,就从877个样本中挑出了80个样本,但是,我们只要这80个样本中成对样本的表达数据,即同一个病人既有癌旁正常细胞的表达蛋白,又有肿瘤细胞的异常表达蛋白。
| submitter_id | n |
|---|---|
| TCGA-50-8457 | 2 |
| TCGA-55-7913 | 2 |
| TCGA-55-8089 | 2 |
| TCGA-44-6148 | 2 |
| TCGA-50-6593 | 2 |
| TCGA-99-8028 | 2 |
| TCGA-50-8459 | 2 |
| TCGA-50-6594 | 2 |
| TCGA-50-5936 | 2 |
| TCGA-55-8205 | 2 |
| TCGA-49-6744 | 2 |
| TCGA-50-8460 | 2 |
| TCGA-49-6742 | 2 |
| TCGA-99-8033 | 2 |
| TCGA-49-6745 | 2 |
| TCGA-91-6848 | 2 |
| TCGA-50-5066 | 2 |
| TCGA-50-5935 | 2 |
| TCGA-J2-8192 | 2 |
| TCGA-44-6145 | 2 |
| TCGA-55-7914 | 2 |
sample <- left_join(PairedSample,pf,by=join_by("submitter_id"))
sample| submitter_id | n | submitter_id.samples | radiation_therapy | kras_mutation_found |
|---|---|---|---|---|
| TCGA-50-8457 | 2 | TCGA-50-8457-01A | NO | YES |
| TCGA-50-8457 | 2 | TCGA-50-8457-11A | NO | YES |
| TCGA-55-7913 | 2 | TCGA-55-7913-01B | NO | NO |
| TCGA-55-7913 | 2 | TCGA-55-7913-11A | NO | NO |
| TCGA-55-8089 | 2 | TCGA-55-8089-01A | NO | NO |
| TCGA-55-8089 | 2 | TCGA-55-8089-11A | NO | NO |
| TCGA-44-6148 | 2 | TCGA-44-6148-01A | NO | NO |
| TCGA-44-6148 | 2 | TCGA-44-6148-11A | NO | NO |
| TCGA-50-6593 | 2 | TCGA-50-6593-01A | YES | NO |
| TCGA-50-6593 | 2 | TCGA-50-6593-11A | YES | NO |
| TCGA-99-8028 | 2 | TCGA-99-8028-01A | NO | YES |
| TCGA-99-8028 | 2 | TCGA-99-8028-11A | NO | YES |
| TCGA-50-8459 | 2 | TCGA-50-8459-01A | NO | YES |
| TCGA-50-8459 | 2 | TCGA-50-8459-11A | NO | YES |
| TCGA-50-6594 | 2 | TCGA-50-6594-01A | NO | NO |
| TCGA-50-6594 | 2 | TCGA-50-6594-11A | NO | NO |
| TCGA-50-5936 | 2 | TCGA-50-5936-01A | NO | YES |
| TCGA-50-5936 | 2 | TCGA-50-5936-11A | NO | YES |
| TCGA-55-8205 | 2 | TCGA-55-8205-01A | NO | NO |
| TCGA-55-8205 | 2 | TCGA-55-8205-11A | NO | NO |
| TCGA-49-6744 | 2 | TCGA-49-6744-01A | NO | YES |
| TCGA-49-6744 | 2 | TCGA-49-6744-11A | NO | YES |
| TCGA-50-8460 | 2 | TCGA-50-8460-01A | YES | NO |
| TCGA-50-8460 | 2 | TCGA-50-8460-11A | YES | NO |
| TCGA-49-6742 | 2 | TCGA-49-6742-01A | NO | NO |
| TCGA-49-6742 | 2 | TCGA-49-6742-11A | NO | NO |
| TCGA-99-8033 | 2 | TCGA-99-8033-01A | YES | NO |
| TCGA-99-8033 | 2 | TCGA-99-8033-11A | YES | NO |
| TCGA-49-6745 | 2 | TCGA-49-6745-01A | NO | NO |
| TCGA-49-6745 | 2 | TCGA-49-6745-11A | NO | NO |
| TCGA-91-6848 | 2 | TCGA-91-6848-01A | NO | NO |
| TCGA-91-6848 | 2 | TCGA-91-6848-11A | NO | NO |
| TCGA-50-5066 | 2 | TCGA-50-5066-01A | NO | NO |
| TCGA-50-5066 | 2 | TCGA-50-5066-02A | NO | NO |
| TCGA-50-5935 | 2 | TCGA-50-5935-01A | YES | YES |
| TCGA-50-5935 | 2 | TCGA-50-5935-11A | YES | YES |
| TCGA-J2-8192 | 2 | TCGA-J2-8192-01A | NO | NO |
| TCGA-J2-8192 | 2 | TCGA-J2-8192-11A | NO | NO |
| TCGA-44-6145 | 2 | TCGA-44-6145-01A | NO | NO |
| TCGA-44-6145 | 2 | TCGA-44-6145-11A | NO | NO |
| TCGA-55-7914 | 2 | TCGA-55-7914-01A | NO | YES |
| TCGA-55-7914 | 2 | TCGA-55-7914-11A | NO | YES |
table(sample$radiation_therapy,sample$kras_mutation_found)
#>
#> NO YES
#> NO 22 12
#> YES 6 2并且colData 中出现的成对样本(21对)要匹配到assays 中的成对样本(60对)。(取交集)
取sample和paired_patient的交集,有8对成对数据
# 取交集
final_sample <- lubridate::intersect(sample$submitter_id,paired_patient)
final_sample
#> [1] "TCGA-44-6148" "TCGA-50-5936" "TCGA-49-6744" "TCGA-49-6742" "TCGA-49-6745"
#> [6] "TCGA-50-5066" "TCGA-50-5935" "TCGA-44-6145"
sample <- sample |>
dplyr::filter(sample$submitter_id %in% final_sample) |>
dplyr::select(-submitter_id, -n) |>
dplyr::rename("sample_id" = "submitter_id.samples") |>
arrange(sample_id)
sample| sample_id | radiation_therapy | kras_mutation_found |
|---|---|---|
| TCGA-44-6145-01A | NO | NO |
| TCGA-44-6145-11A | NO | NO |
| TCGA-44-6148-01A | NO | NO |
| TCGA-44-6148-11A | NO | NO |
| TCGA-49-6742-01A | NO | NO |
| TCGA-49-6742-11A | NO | NO |
| TCGA-49-6744-01A | NO | YES |
| TCGA-49-6744-11A | NO | YES |
| TCGA-49-6745-01A | NO | NO |
| TCGA-49-6745-11A | NO | NO |
| TCGA-50-5066-01A | NO | NO |
| TCGA-50-5066-02A | NO | NO |
| TCGA-50-5935-01A | YES | YES |
| TCGA-50-5935-11A | YES | YES |
| TCGA-50-5936-01A | NO | YES |
| TCGA-50-5936-11A | NO | YES |
table(sample$radiation_therapy,sample$kras_mutation_found)
#>
#> NO YES
#> NO 10 4
#> YES 0 2
# 最后的表达矩阵
exprCounts <- Counts |> dplyr::select(which(colnames(Counts) %in% sample$sample_id))
exprCounts <- dplyr::select(exprCounts,order(colnames(exprCounts)))
colnames(exprCounts)
#> [1] "TCGA-44-6145-01A" "TCGA-44-6145-11A" "TCGA-44-6148-01A" "TCGA-44-6148-11A"
#> [5] "TCGA-49-6742-01A" "TCGA-49-6742-11A" "TCGA-49-6744-01A" "TCGA-49-6744-11A"
#> [9] "TCGA-49-6745-01A" "TCGA-49-6745-11A" "TCGA-50-5066-01A" "TCGA-50-5066-02A"
#> [13] "TCGA-50-5935-01A" "TCGA-50-5935-11A" "TCGA-50-5936-01A" "TCGA-50-5936-11A"
dim(exprCounts)
#> [1] 58387 16
# 去除所有样本都不表达的基因
exprCounts <- exprCounts |>
mutate(rowsum = apply(exprCounts,1,sum)) |>
dplyr::filter(rowsum!=0) |>
dplyr::select(-rowsum)
dim(exprCounts)
#> [1] 46488 16# 检查计数矩阵和列数据,看看它们在样本顺序方面是否一致。
colnames(exprCounts)
#> [1] "TCGA-44-6145-01A" "TCGA-44-6145-11A" "TCGA-44-6148-01A" "TCGA-44-6148-11A"
#> [5] "TCGA-49-6742-01A" "TCGA-49-6742-11A" "TCGA-49-6744-01A" "TCGA-49-6744-11A"
#> [9] "TCGA-49-6745-01A" "TCGA-49-6745-11A" "TCGA-50-5066-01A" "TCGA-50-5066-02A"
#> [13] "TCGA-50-5935-01A" "TCGA-50-5935-11A" "TCGA-50-5936-01A" "TCGA-50-5936-11A"
sample$sample_id
#> [1] "TCGA-44-6145-01A" "TCGA-44-6145-11A" "TCGA-44-6148-01A" "TCGA-44-6148-11A"
#> [5] "TCGA-49-6742-01A" "TCGA-49-6742-11A" "TCGA-49-6744-01A" "TCGA-49-6744-11A"
#> [9] "TCGA-49-6745-01A" "TCGA-49-6745-11A" "TCGA-50-5066-01A" "TCGA-50-5066-02A"
#> [13] "TCGA-50-5935-01A" "TCGA-50-5935-11A" "TCGA-50-5936-01A" "TCGA-50-5936-11A"
all(sample$sample_id%in% colnames(exprCounts))
#> [1] TRUE
all(sample$SampleID == colnames(exprCounts))
#> [1] TRUE根据TCGA样本的命名可以区分正常组织和肿瘤样本的测序结果 其中编号01-09表示肿瘤,10-19表示正常
# 字符14~15,
sample$condition <- factor(
ifelse(as.numeric(str_sub(sample$sample_id,14,15)) < 10,'tumor','normal'),
levels = c("normal","tumor"),
)
sample <- column_to_rownames(sample,var ="sample_id" )
sample| radiation_therapy | kras_mutation_found | condition | |
|---|---|---|---|
| TCGA-44-6145-01A | NO | NO | tumor |
| TCGA-44-6145-11A | NO | NO | normal |
| TCGA-44-6148-01A | NO | NO | tumor |
| TCGA-44-6148-11A | NO | NO | normal |
| TCGA-49-6742-01A | NO | NO | tumor |
| TCGA-49-6742-11A | NO | NO | normal |
| TCGA-49-6744-01A | NO | YES | tumor |
| TCGA-49-6744-11A | NO | YES | normal |
| TCGA-49-6745-01A | NO | NO | tumor |
| TCGA-49-6745-11A | NO | NO | normal |
| TCGA-50-5066-01A | NO | NO | tumor |
| TCGA-50-5066-02A | NO | NO | tumor |
| TCGA-50-5935-01A | YES | YES | tumor |
| TCGA-50-5935-11A | YES | YES | normal |
| TCGA-50-5936-01A | NO | YES | tumor |
| TCGA-50-5936-11A | NO | YES | normal |
# BiocManager::install("DESeq2")
library(DESeq2)
conflicts_prefer(GenomicRanges::setdiff)
dds <- DESeqDataSetFromMatrix(countData = exprCounts,
colData = sample,
design = ~ condition)
dds
#> class: DESeqDataSet
#> dim: 46488 16
#> metadata(1): version
#> assays(1): counts
#> rownames(46488): TSPAN6 TNMD ... LINC01144 RP11-418H16.1
#> rowData names(0):
#> colnames(16): TCGA-44-6145-01A TCGA-44-6145-11A ... TCGA-50-5936-01A
#> TCGA-50-5936-11A
#> colData names(3): radiation_therapy kras_mutation_found conditionsmallestGroupSize <- 8
keep <- rowSums(counts(dds) >= 10) >= smallestGroupSize
dds <- dds[keep,]
dds
#> class: DESeqDataSet
#> dim: 19562 16
#> metadata(1): version
#> assays(1): counts
#> rownames(19562): TSPAN6 DPM1 ... RP11-274B21.13 LINC01144
#> rowData names(0):
#> colnames(16): TCGA-44-6145-01A TCGA-44-6145-11A ... TCGA-50-5936-01A
#> TCGA-50-5936-11A
#> colData names(3): radiation_therapy kras_mutation_found conditiondds$condition
#> [1] tumor normal tumor normal tumor normal tumor normal tumor normal
#> [11] tumor tumor tumor normal tumor normal
#> Levels: normal tumor
dds@colData
#> DataFrame with 16 rows and 3 columns
#> radiation_therapy kras_mutation_found condition
#> <character> <character> <factor>
#> TCGA-44-6145-01A NO NO tumor
#> TCGA-44-6145-11A NO NO normal
#> TCGA-44-6148-01A NO NO tumor
#> TCGA-44-6148-11A NO NO normal
#> TCGA-49-6742-01A NO NO tumor
#> ... ... ... ...
#> TCGA-50-5066-02A NO NO tumor
#> TCGA-50-5935-01A YES YES tumor
#> TCGA-50-5935-11A YES YES normal
#> TCGA-50-5936-01A NO YES tumor
#> TCGA-50-5936-11A NO YES normal
head(counts(dds)) # 19562 × 16
#> TCGA-44-6145-01A TCGA-44-6145-11A TCGA-44-6148-01A TCGA-44-6148-11A
#> TSPAN6 1407 783 4794 3163
#> DPM1 1293 1003 982 1158
#> SCYL3 758 389 985 885
#> C1orf112 340 74 173 183
#> FGR 747 3617 759 3076
#> CFH 5718 2611 30270 8703
#> TCGA-49-6742-01A TCGA-49-6742-11A TCGA-49-6744-01A TCGA-49-6744-11A
#> TSPAN6 3275 1696 2798 1360
#> DPM1 922 986 1453 846
#> SCYL3 838 498 808 514
#> C1orf112 261 95 271 119
#> FGR 286 2126 1470 2712
#> CFH 1574 5208 5880 3922
#> TCGA-49-6745-01A TCGA-49-6745-11A TCGA-50-5066-01A TCGA-50-5066-02A
#> TSPAN6 2700 2194 3589 8759
#> DPM1 1159 1584 2848 4398
#> SCYL3 555 570 602 2118
#> C1orf112 386 98 483 1518
#> FGR 1065 1631 541 1169
#> CFH 6554 4153 2071 9081
#> TCGA-50-5935-01A TCGA-50-5935-11A TCGA-50-5936-01A TCGA-50-5936-11A
#> TSPAN6 2199 1202 2537 725
#> DPM1 1129 715 621 769
#> SCYL3 1349 441 556 553
#> C1orf112 386 60 346 80
#> FGR 560 1831 519 3600
#> CFH 1869 3667 4488 1881des <- DESeq(dds)
results <- results(
object = des,
contrast = c("condition", "tumor", "normal"),
alpha = 0.05,
filter = rowMeans(counts(des, normalized = TRUE)),# 独立过滤
theta = c(0.025, 0.975),
pAdjustMethod = "BH",
)
metadata(results)["alpha"]
#> $alpha
#> [1] 0.05
results[1:6,]
#> log2 fold change (MLE): condition tumor vs normal
#> Wald test p-value: condition tumor vs normal
#> DataFrame with 6 rows and 6 columns
#> baseMean log2FoldChange lfcSE stat pvalue padj
#> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
#> TSPAN6 2491.784 1.009957 0.284407 3.55110 3.83631e-04 2.72195e-03
#> DPM1 1318.670 0.500631 0.336588 1.48737 1.36918e-01 2.72805e-01
#> SCYL3 734.633 0.605618 0.202489 2.99087 2.78184e-03 1.38997e-02
#> C1orf112 268.645 1.951846 0.287025 6.80026 1.04432e-11 7.96688e-10
#> FGR 1655.039 -1.947828 0.290586 -6.70310 2.04049e-11 1.44134e-09
#> CFH 5564.337 0.608888 0.464523 1.31078 1.89932e-01 3.45383e-01
summary(results)
#>
#> out of 19562 with nonzero total read count
#> adjusted p-value < 0.05
#> LFC > 0 (up) : 3248, 17%
#> LFC < 0 (down) : 2160, 11%
#> outliers [1] : 0, 0%
#> low counts [2] : 490, 2.5%
#> (mean count < 13)
#> [1] see 'cooksCutoff' argument of ?results
#> [2] see 'independentFiltering' argument of ?results
sum(results$padj < 0.05, na.rm=TRUE)
#> [1] 5408
# 根据padj 升序
resOrdered <- results[order(results$padj),]保存csv以备热图、火山图、富集分析。
x <- as.data.frame(resOrdered) |> rownames_to_column(var = "gene")
write_csv(x, file="data/DEGs_Analysis/resOrdered.csv")mcols(resOrdered)$description
#> [1] "mean of normalized counts for all samples"
#> [2] "log2 fold change (MLE): condition tumor vs normal"
#> [3] "standard error: condition tumor vs normal"
#> [4] "Wald statistic: condition tumor vs normal"
#> [5] "Wald test p-value: condition tumor vs normal"
#> [6] "BH adjusted p-values"pvalue,是统计学检验变量,代表差异显著性,一般认为P < 0.05 为显著, P <0.01 为非常显著。其含义为:由抽样误差导致样本间差异的概率小于0.05 或0.01。
padj,是对pvalue进行多重假设检验校正后的结果。转录组测序的差异表达分析是对大量的基因表达值进行的独立统计假设检验,存在假阳性(Family-Wise Error Rate,FWER),Bonferroni矫正
qvalue,False Discovery Rate,FDR,所有假阳性结果占所有拒绝零假设结果的比例,假阳性结果的比例不超过预设的FDR水平(0.05/0.1),基于pvalue的分布。多重假设检验校正方法:Benjamini-Hochberg,
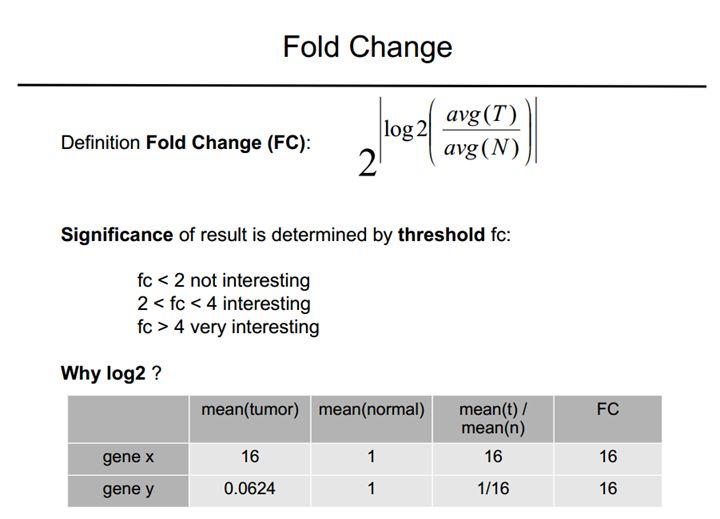
log2FoldChange:对数倍数变化,1.5倍差异即0.585,2倍差异即1。
DEG <- read_csv("data/DEGs_Analysis/resOrdered.csv")
DESeq2_DEG <- na.omit(DEG)
nrDEG <- DESeq2_DEG
nrDEG[1:25,]| gene | baseMean | log2FoldChange | lfcSE | stat | pvalue | padj |
|---|---|---|---|---|---|---|
| MGAT3 | 1412.43862 | -4.320278 | 0.2750470 | -15.707414 | 0 | 0 |
| RTKN2 | 5130.81804 | -4.640307 | 0.3175103 | -14.614667 | 0 | 0 |
| GPM6A | 1228.70589 | -5.145635 | 0.3599700 | -14.294621 | 0 | 0 |
| SERTM1 | 298.17214 | -6.886260 | 0.5455626 | -12.622309 | 0 | 0 |
| LIMS2 | 1926.05684 | -2.660279 | 0.2146619 | -12.392878 | 0 | 0 |
| EMP2 | 31710.06920 | -2.671839 | 0.2229468 | -11.984199 | 0 | 0 |
| CNTN6 | 352.36029 | -4.393899 | 0.3796112 | -11.574736 | 0 | 0 |
| KANK3 | 495.11283 | -2.848364 | 0.2522794 | -11.290513 | 0 | 0 |
| RP1-78O14.1 | 240.67218 | -4.394661 | 0.3899644 | -11.269390 | 0 | 0 |
| GPM6B | 796.26123 | -2.528567 | 0.2297754 | -11.004520 | 0 | 0 |
| WNT3A | 280.08606 | -5.270964 | 0.4854622 | -10.857621 | 0 | 0 |
| PDLIM2 | 1757.53896 | -2.207799 | 0.2064731 | -10.692915 | 0 | 0 |
| ITLN2 | 655.86747 | -5.904468 | 0.5588234 | -10.565893 | 0 | 0 |
| SLC6A13 | 35.46976 | -3.659195 | 0.3472796 | -10.536740 | 0 | 0 |
| GRK5 | 2389.94916 | -2.795107 | 0.2712779 | -10.303480 | 0 | 0 |
| EDNRB | 3236.38793 | -3.288481 | 0.3211332 | -10.240242 | 0 | 0 |
| KCNN4 | 1240.55071 | 3.836884 | 0.3783283 | 10.141679 | 0 | 0 |
| CLIC5 | 8327.11939 | -3.732657 | 0.3685299 | -10.128505 | 0 | 0 |
| RASAL1 | 261.44562 | 4.538838 | 0.4504928 | 10.075272 | 0 | 0 |
| RP11-371A19.2 | 54.44687 | -5.168047 | 0.5162906 | -10.009957 | 0 | 0 |
| PTPN21 | 2327.21817 | -2.527771 | 0.2538084 | -9.959365 | 0 | 0 |
| CLEC3B | 2091.74017 | -4.223742 | 0.4294719 | -9.834733 | 0 | 0 |
| CTD-2135D7.5 | 38.21341 | -3.366170 | 0.3434858 | -9.800029 | 0 | 0 |
| FAM83A | 2565.85011 | 6.377845 | 0.6514036 | 9.790927 | 0 | 0 |
| PTPRH | 490.58361 | 6.130422 | 0.6281505 | 9.759480 | 0 | 0 |
挑选指定第一个基因看它在同一个病人的配对表达情况
library(ggpubr)
df <- tibble(
group = sample$condition,
patient = colnames(exprCounts),
expressionValue = as.numeric(exprCounts[DESeq2_DEG$gene[1],]),
)
ggpubr::ggpaired(df, x = "group", y = "expressionValue",color = "group",
line.color = "gray", line.size = 0.4,palette = "npg")+
stat_compare_means()
nrDEG$group <- factor(
if_else(nrDEG$padj<0.05& abs(nrDEG$log2FoldChange)>=1,
if_else(nrDEG$log2FoldChange>=1,"up","down"),
"NS",),
levels =c("up","down","NS") )
table(nrDEG$group)
#>
#> up down NS
#> 2073 1641 15358
gene_labels <- dplyr::filter(nrDEG,nrDEG$padj<0.05 &
abs(nrDEG$log2FoldChange)>=1)|>
slice_head(n=25)
ggplot(nrDEG,aes( x = log2FoldChange,y = -log10(padj),color=group,shape=group))+
geom_point(alpha=0.5,size=1.5)+
scale_color_manual(values = c("red","green","gray"))+
#scale_shape_manual(values = c(2,25,4))+
geom_vline(xintercept = c(-1,0,1),lty=3,color="grey25",lwd=0.8)+
geom_hline(yintercept = -log10(0.05),lty=3,color="grey25",lwd=0.8)+
ggrepel::geom_text_repel(
data = gene_labels,
aes(label=gene),
color="black",
size=2)+
theme_pubr()
if(!require(EnhancedVolcano))
BiocManager::install('EnhancedVolcano')resultsNames(des)
#> [1] "Intercept" "condition_tumor_vs_normal"
# BiocManager::install("apeglm") 自适应 t 先验收缩估计器 默认
# resLFC <- lfcShrink(des, coef="condition_tumor_vs_normal", type="apeglm")
# `normal`是原始的 DESeq2 收缩估计器,是先验的自适应正态分布
resNorm <- lfcShrink(des, coef=2, type="normal")
# BiocManager::install("ashr")
# resAsh <- lfcShrink(des, coef=2, type="ashr")
## 先验信息
# priorInfo(resLFC)
priorInfo(resNorm)
#> $type
#> [1] "normal"
#>
#> $package
#> [1] "DESeq2"
#>
#> $version
#> [1] '1.44.0'
#>
#> $betaPriorVar
#> Intercept conditiontumor
#> 1.000000e+06 1.658755e+00
# priorInfo(resAsh)# 对gene添加“是否呈现显著差异表达”的标签
DESeq2_DEG$significant <- factor(ifelse(DESeq2_DEG$padj <0.05, "Significant", "NS"),
levels = c("Significant", "NS"))
table(DESeq2_DEG$significant)
#>
#> Significant NS
#> 5408 13664
# 以baseMean作为x,log2FoldChange作为y 以significant作为分类变量
ggplot(DESeq2_DEG, aes(baseMean, log2FoldChange, colour=significant)) +
geom_point(size=1) +
scale_y_continuous(limits=c(-3, 3)) +
scale_x_log10() +
geom_hline(yintercept = 0, colour="black", linewidth=1) +
labs(x="mean of normalized counts", y="log2FoldChange") +
scale_colour_manual(name="padj",
values=c("Significant"="blue","NS"="grey50")) +
theme_classic()
# the counts of reads for a single gene across the groups
plotCounts(des, gene=which.min(resOrdered$padj),
intgroup="condition",returnData=TRUE) |>
ggplot(aes(x=condition, y=count)) +
geom_point(position=position_jitter(w=0.1,h=0))
colData(dds)
#> DataFrame with 16 rows and 3 columns
#> radiation_therapy kras_mutation_found condition
#> <character> <character> <factor>
#> TCGA-44-6145-01A NO NO tumor
#> TCGA-44-6145-11A NO NO normal
#> TCGA-44-6148-01A NO NO tumor
#> TCGA-44-6148-11A NO NO normal
#> TCGA-49-6742-01A NO NO tumor
#> ... ... ... ...
#> TCGA-50-5066-02A NO NO tumor
#> TCGA-50-5935-01A YES YES tumor
#> TCGA-50-5935-11A YES YES normal
#> TCGA-50-5936-01A NO YES tumor
#> TCGA-50-5936-11A NO YES normal


