1 上游分析
1.1 测序
1.1.1 FASTQ 文件

@<instrument>:<run num>:<flowcell ID>:<lane>:<tile>:<x>:<y>:<UMI> <read>:<filtered>:<control num>:<index>
Fastq文件形如:
@M00970:337:000000000-BR5KF:1:1102:17745:1557 1:N:0:CGCAGAAC+ACAGAGTT
ACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCT...AAACGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGC
+
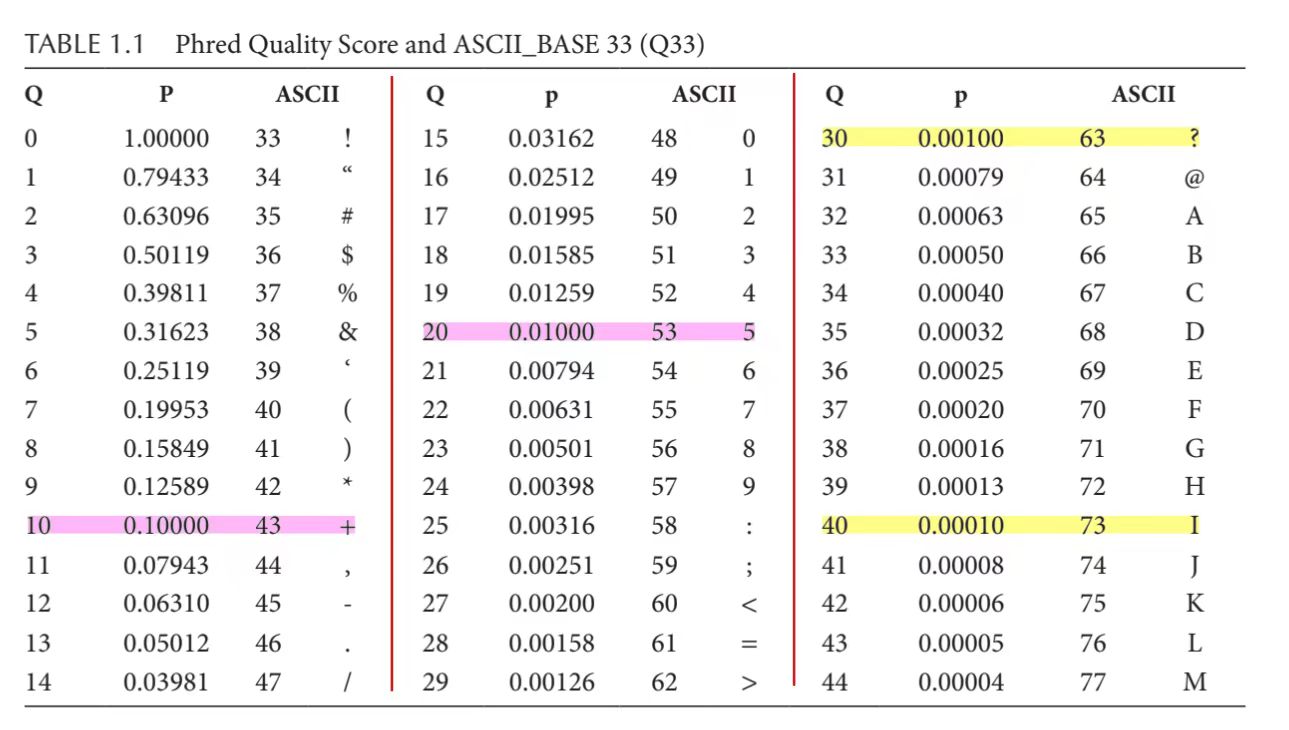
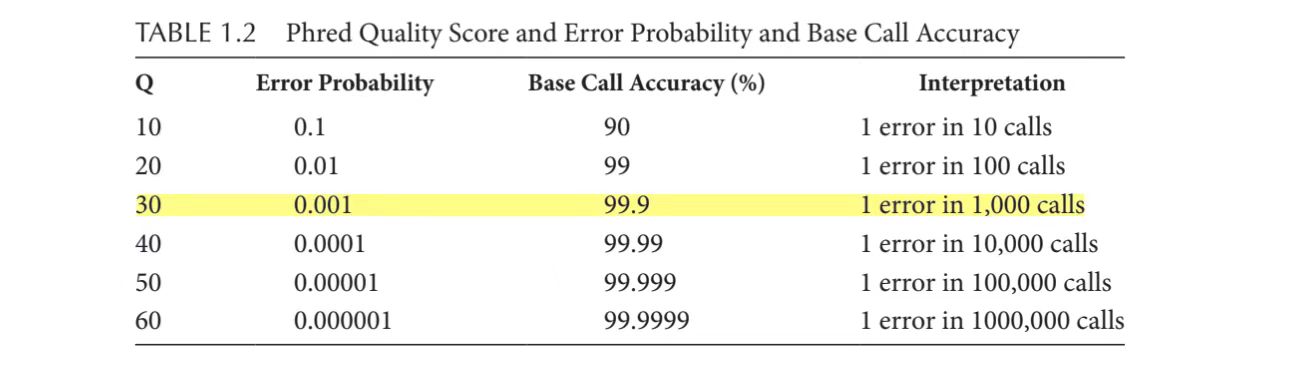
DDDCDDDDDDFFGGGGGGGGGGHHHHHHHHHHHHHHH...HHHHGGGGHHHHHHHHHHHHHHHGHHGHHHHHHGGGGGGHHHH每个碱基对应的字符在ASCII码中对应的十进制数字减去33即为该碱基质量(也即Phred33体系),例如上述序列中第一个碱基的质量为D,对应的十进制数字为68(见下表),则碱基质量为68-33=35。碱基质量Q=-10*lgP,P为碱基被测错的概率。也即Q为30代表被测错的概率为0.001,碱基质量越高,则被测错的概率越低。
Q = -10 log10(P)
1.1.2 BAM 文件
Binary alignment
1.1.3 VCF 文件
变异注释
VariantAnnotation readVcf()
1.1.4 BED、WIG、GTF 文件
基因组注释文件
1.2 质量控制:FastQC
1.3 剪切:Trimmomatic
接头序列的去除和低质量序列的修剪
1.4 序列比对:STAR
1.5 定量:计数矩阵
https://subread.sourceforge.net/featureCounts.html
将读数与基因组对齐,并计算每个基因外显子内的读数数量。
R 包 Rsubread 更简单、更快、更便宜、更适合 RNA 测序的比对和定量
https://bioconductor.org/packages/release/bioc/html/Rsubread.html
featureCount是subread软件包里的一个命令,所以安装subread R版,
将读数与转录组对齐,量化转录表达,并将转录表达总结为基因表达
Pseudoalign针对转录组进行读取,使用相应的基因组作为诱饵,量化该过程中的转录物表达,并将转录物水平的表达总结为基因水平的表达