5 SummarizedExperiment
SummarizeExperiment 如 Figure 5.1 所示
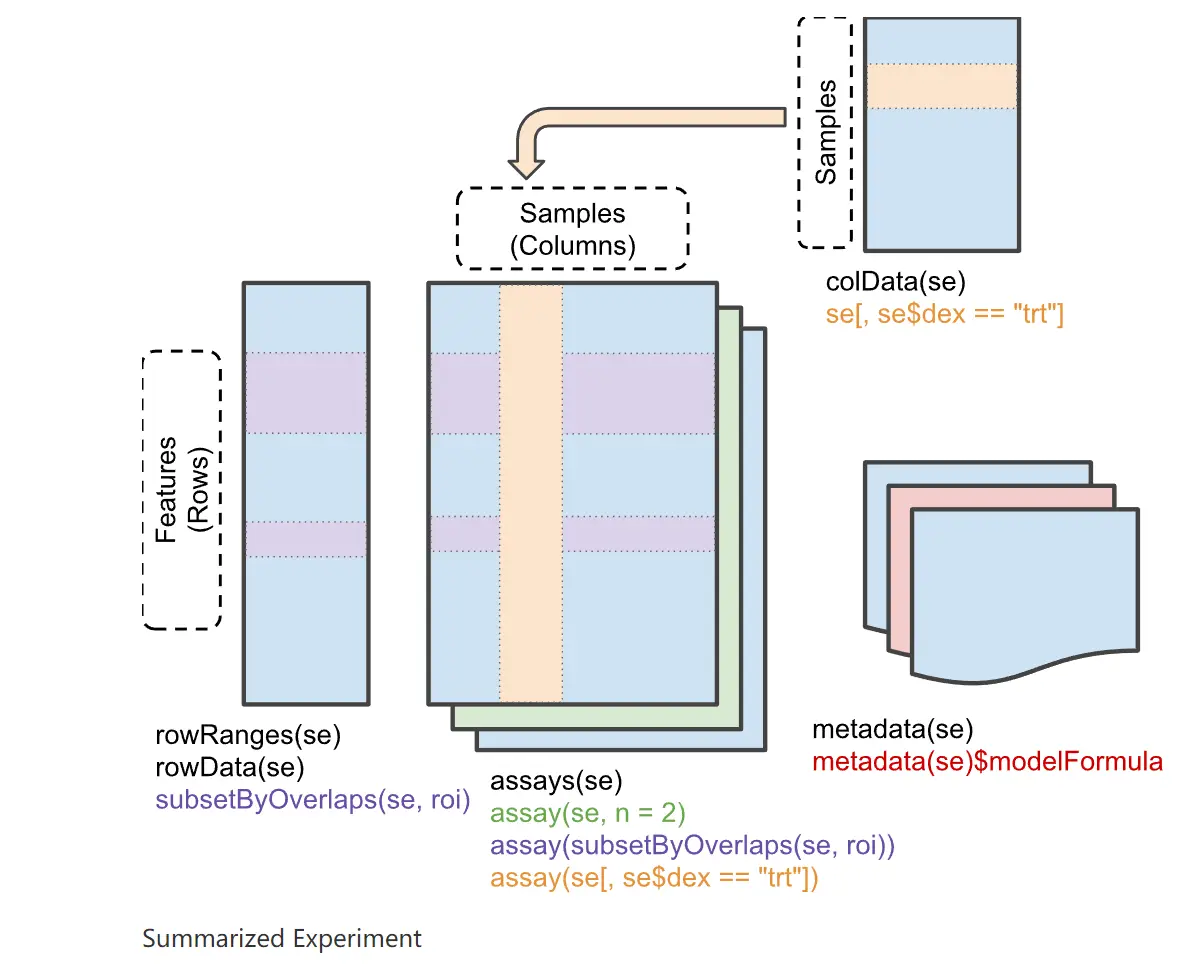
具体可参考 SummarizedExperiment docs 。
5.1 构造SummarizedExperiment实例
Code
# 计数矩阵
nrows <- 200
ncols <- 6
counts <- matrix(runif(nrows * ncols, 1, 1e4), nrows)
head(counts)
#> [,1] [,2] [,3] [,4] [,5] [,6]
#> [1,] 9716.5615 5785.166 6787.2718 9230.371 2980.809 6003.592
#> [2,] 526.7021 7399.303 4174.6909 8129.570 7930.471 9444.718
#> [3,] 3638.8988 5585.446 3146.2578 1827.896 9523.883 1823.681
#> [4,] 4043.9146 9632.294 606.9306 8414.168 9299.789 1467.223
#> [5,] 144.7185 5088.270 8735.6897 5399.352 8930.863 9889.022
#> [6,] 3421.2014 8510.405 9424.0854 9902.199 3764.500 7555.098
# 基因元数据
rowData <- GRanges(seqnames = rep(c("chr1", "chr2"), c(50, 150)),
ranges = IRanges(floor(runif(200, 1e5, 1e6)), width=100),
strand=sample(c("+", "-"), 200, TRUE),
gene_id=sprintf("ID%03d", 1:200))
rowData[1:6,]
#> GRanges object with 6 ranges and 1 metadata column:
#> seqnames ranges strand | gene_id
#> <Rle> <IRanges> <Rle> | <character>
#> [1] chr1 842135-842234 + | ID001
#> [2] chr1 893167-893266 - | ID002
#> [3] chr1 143410-143509 - | ID003
#> [4] chr1 389155-389254 + | ID004
#> [5] chr1 837386-837485 + | ID005
#> [6] chr1 383201-383300 - | ID006
#> -------
#> seqinfo: 2 sequences from an unspecified genome; no seqlengths
# 样本元数据
colData <- DataFrame(Treatment=rep(c("ChIP", "Input"), 3),
row.names=LETTERS[1:6])
colData
#> DataFrame with 6 rows and 1 column
#> Treatment
#> <character>
#> A ChIP
#> B Input
#> C ChIP
#> D Input
#> E ChIP
#> F Input
# 实验元数据
metadata <- "A example of how to create an instance of SummarizedExperiment"
se <- SummarizedExperiment(assays=list(counts=counts),
rowRanges=rowData,
colData=colData,
metadata=metadata)
se
#> class: RangedSummarizedExperiment
#> dim: 200 6
#> metadata(1): ''
#> assays(1): counts
#> rownames: NULL
#> rowData names(1): gene_id
#> colnames(6): A B ... E F
#> colData names(1): TreatmentCode
dim(se)
#> [1] 200 6
SummarizedExperiment::assay(se) %>% head()
#> A B C D E F
#> [1,] 9716.5615 5785.166 6787.2718 9230.371 2980.809 6003.592
#> [2,] 526.7021 7399.303 4174.6909 8129.570 7930.471 9444.718
#> [3,] 3638.8988 5585.446 3146.2578 1827.896 9523.883 1823.681
#> [4,] 4043.9146 9632.294 606.9306 8414.168 9299.789 1467.223
#> [5,] 144.7185 5088.270 8735.6897 5399.352 8930.863 9889.022
#> [6,] 3421.2014 8510.405 9424.0854 9902.199 3764.500 7555.098
SummarizedExperiment::rowData(se)
#> DataFrame with 200 rows and 1 column
#> gene_id
#> <character>
#> 1 ID001
#> 2 ID002
#> 3 ID003
#> 4 ID004
#> 5 ID005
#> ... ...
#> 196 ID196
#> 197 ID197
#> 198 ID198
#> 199 ID199
#> 200 ID200
SummarizedExperiment::rowRanges(se)
#> GRanges object with 200 ranges and 1 metadata column:
#> seqnames ranges strand | gene_id
#> <Rle> <IRanges> <Rle> | <character>
#> [1] chr1 842135-842234 + | ID001
#> [2] chr1 893167-893266 - | ID002
#> [3] chr1 143410-143509 - | ID003
#> [4] chr1 389155-389254 + | ID004
#> [5] chr1 837386-837485 + | ID005
#> ... ... ... ... . ...
#> [196] chr2 828198-828297 + | ID196
#> [197] chr2 897222-897321 - | ID197
#> [198] chr2 815142-815241 - | ID198
#> [199] chr2 990951-991050 + | ID199
#> [200] chr2 153508-153607 - | ID200
#> -------
#> seqinfo: 2 sequences from an unspecified genome; no seqlengths
SummarizedExperiment::colData(se)
#> DataFrame with 6 rows and 1 column
#> Treatment
#> <character>
#> A ChIP
#> B Input
#> C ChIP
#> D Input
#> E ChIP
#> F Input