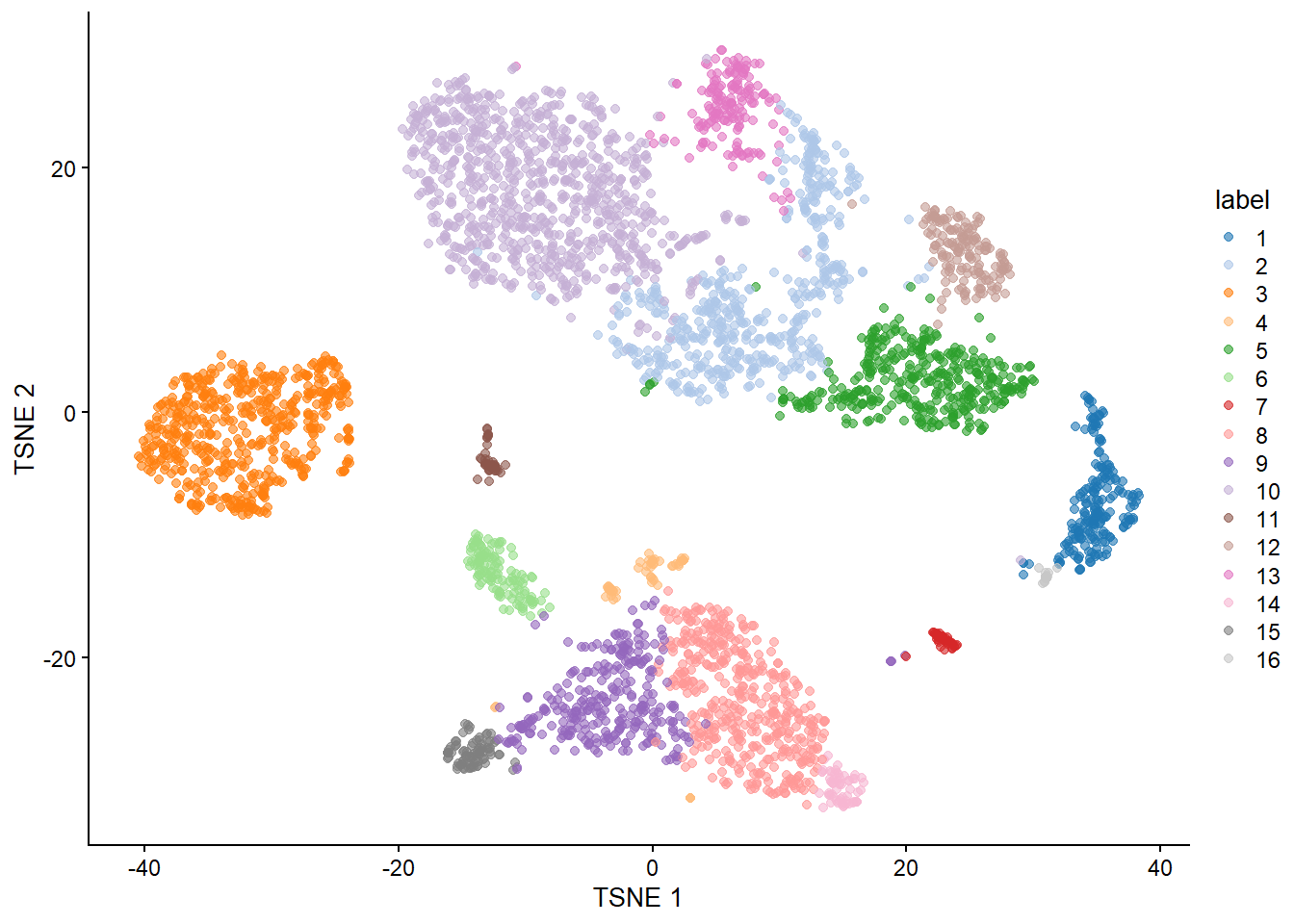
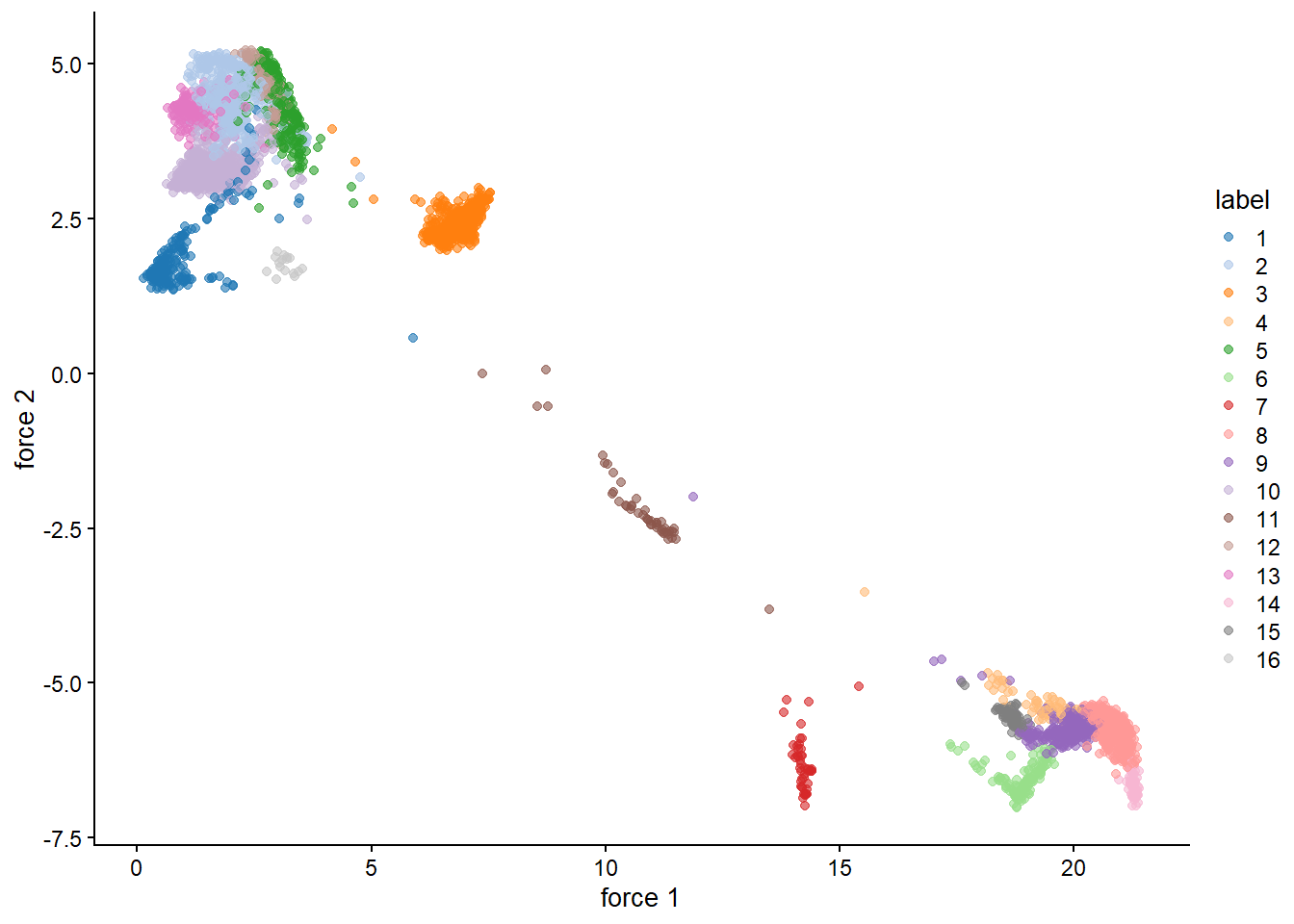
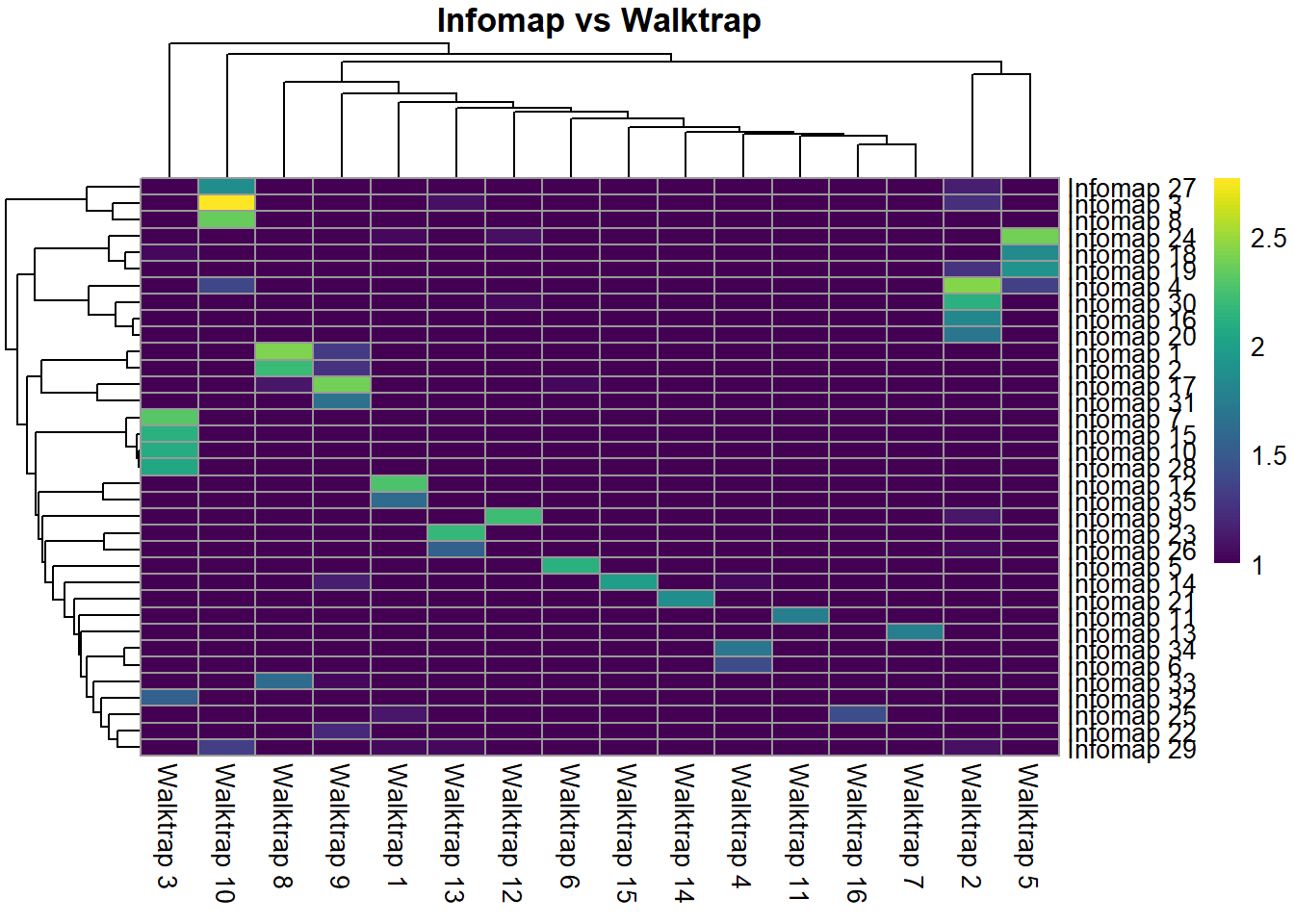
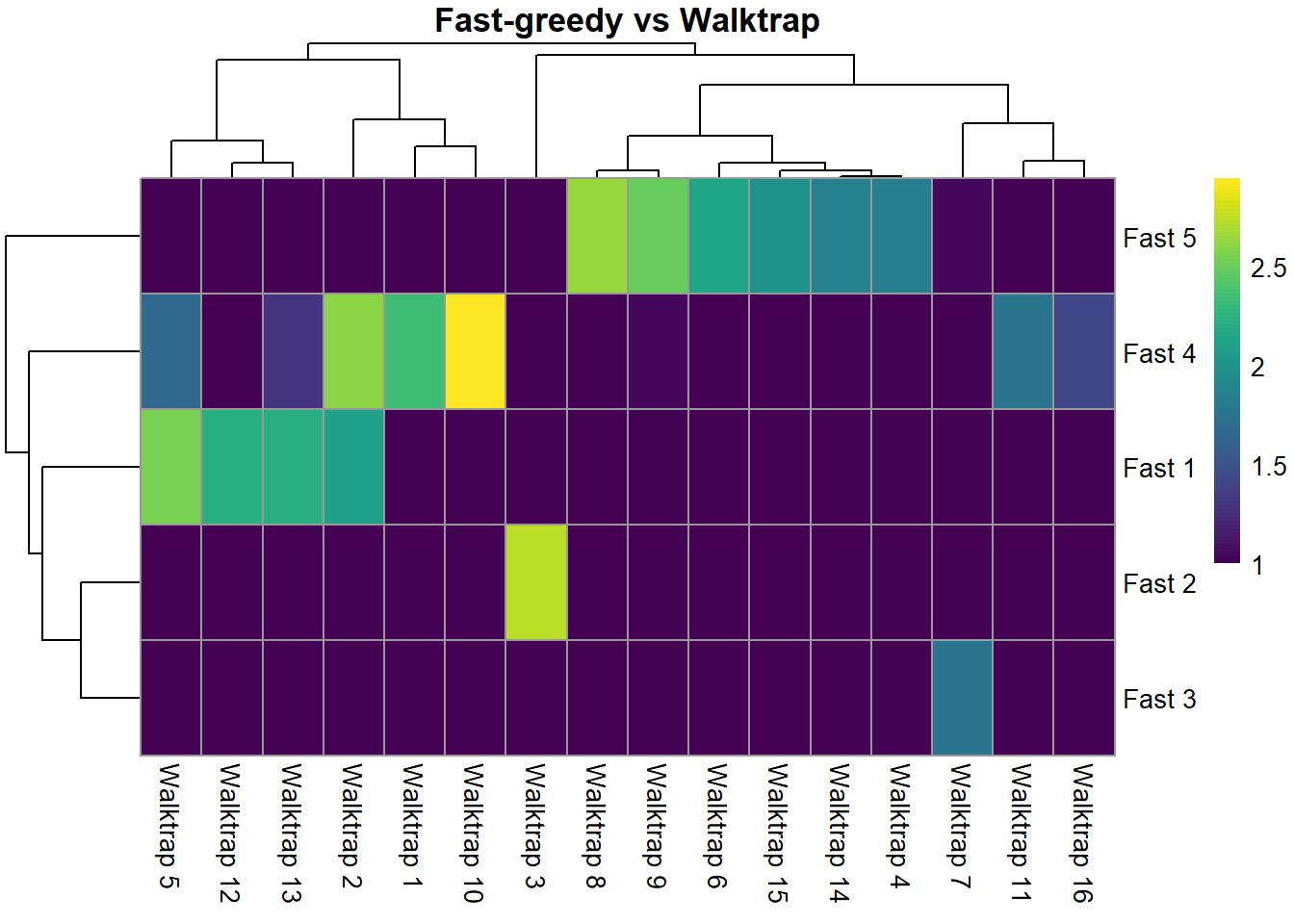
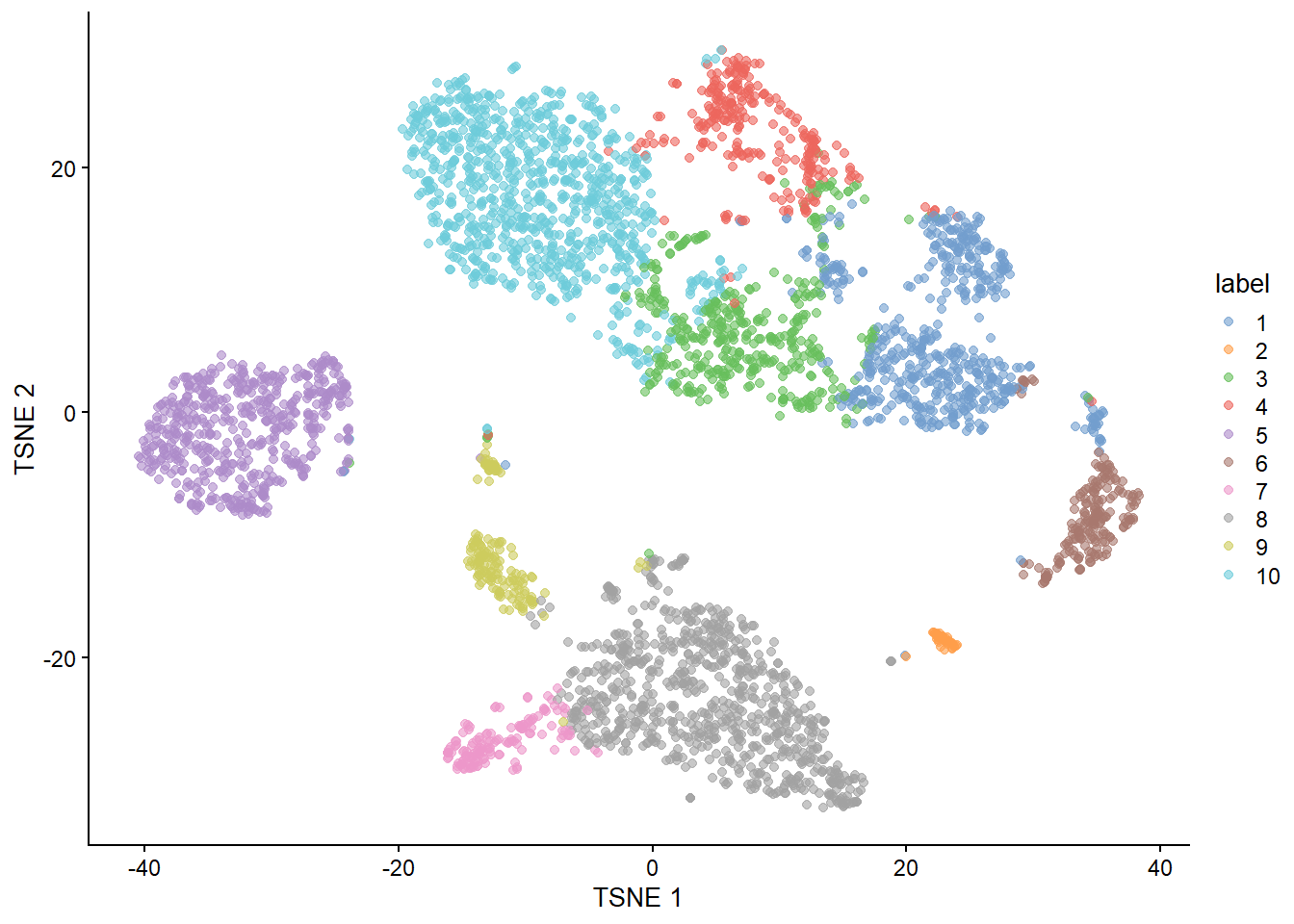
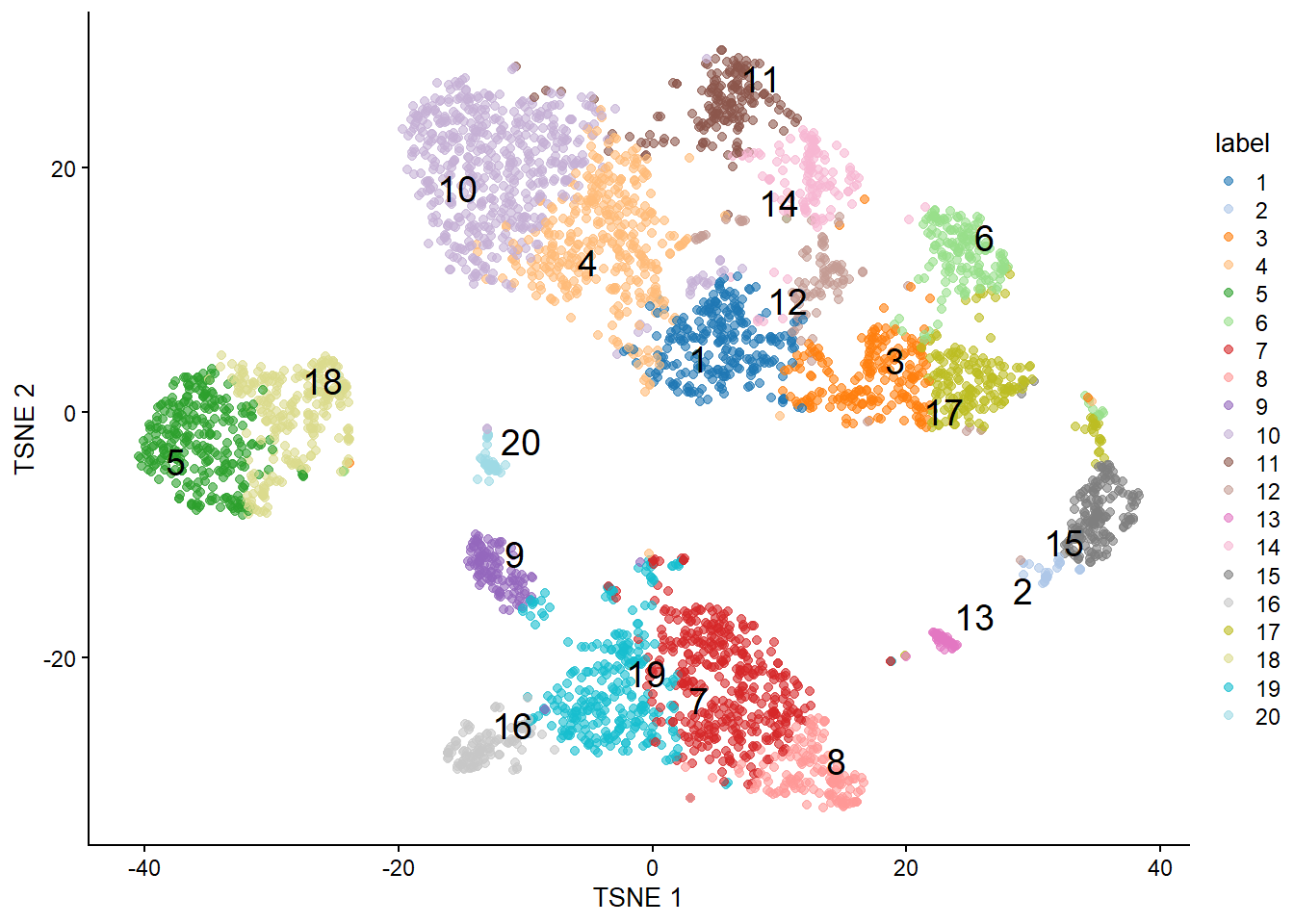
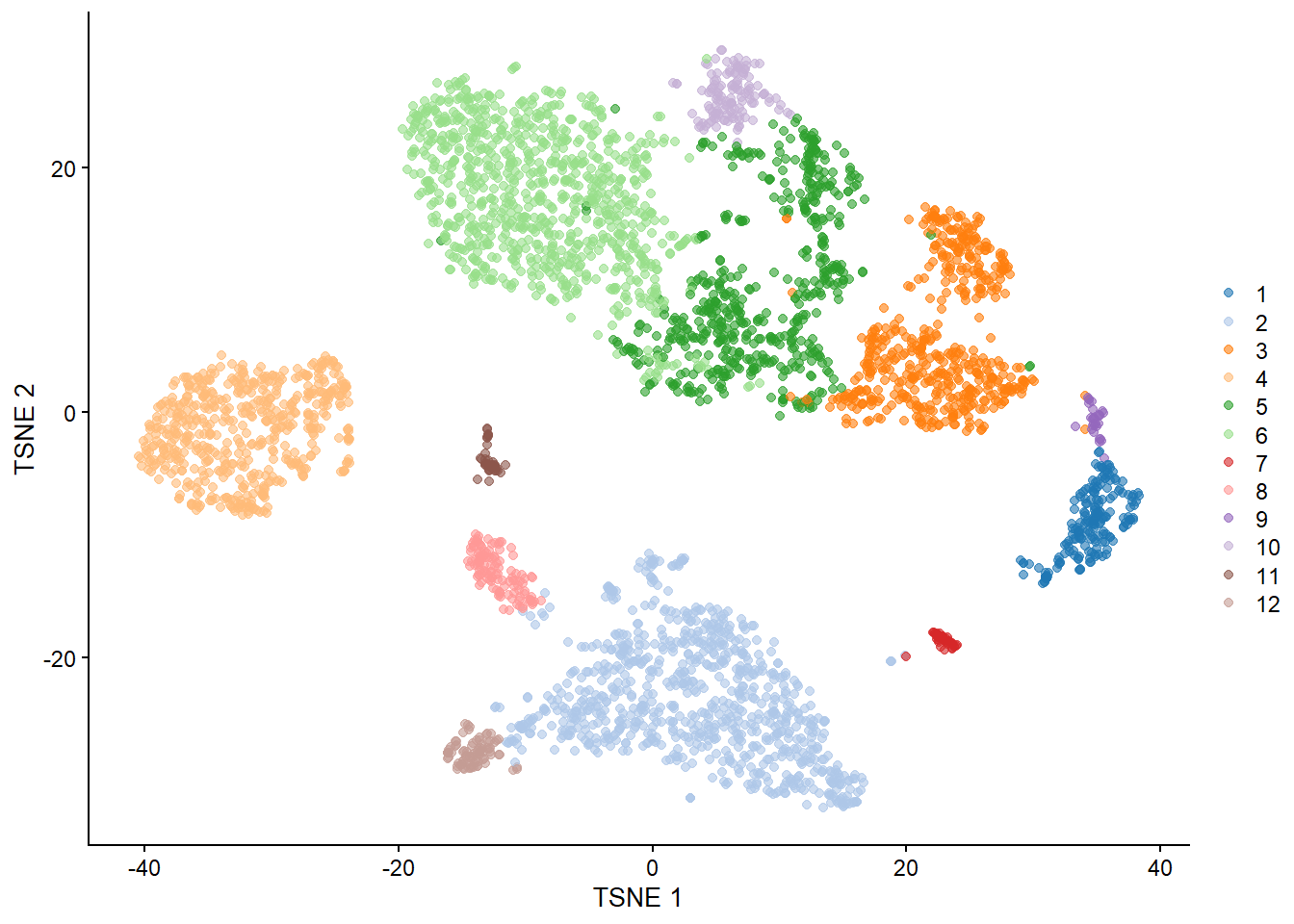
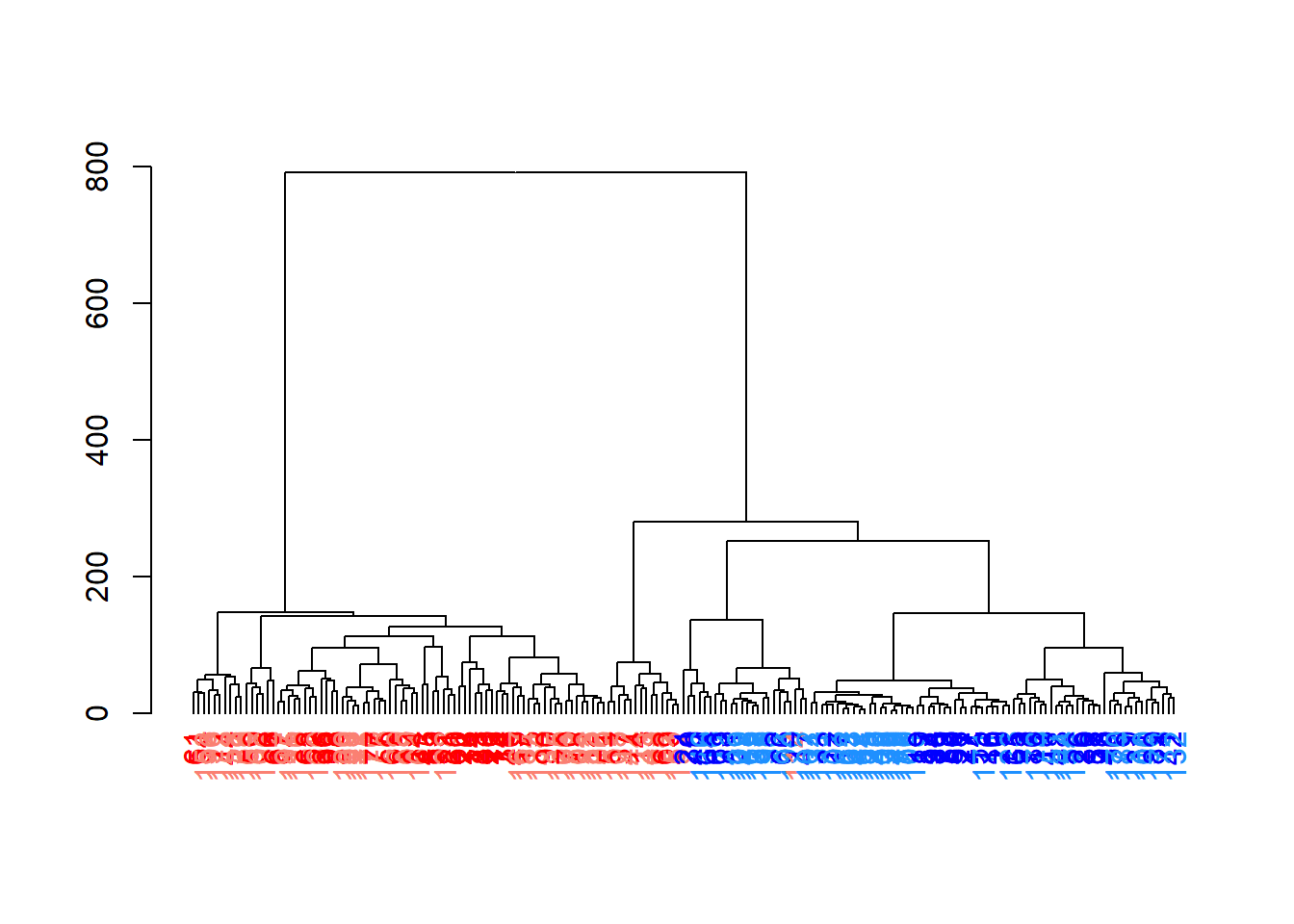
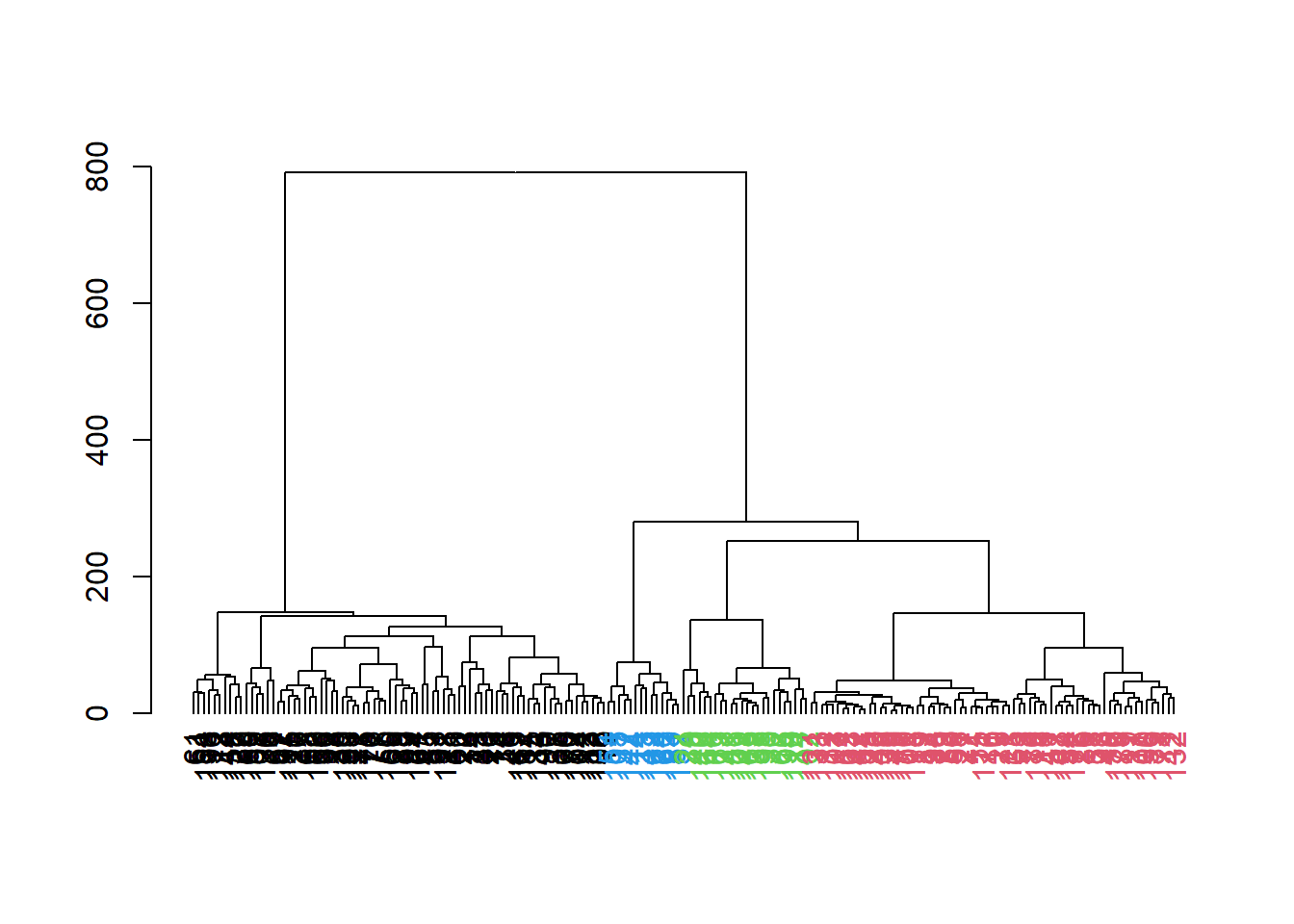
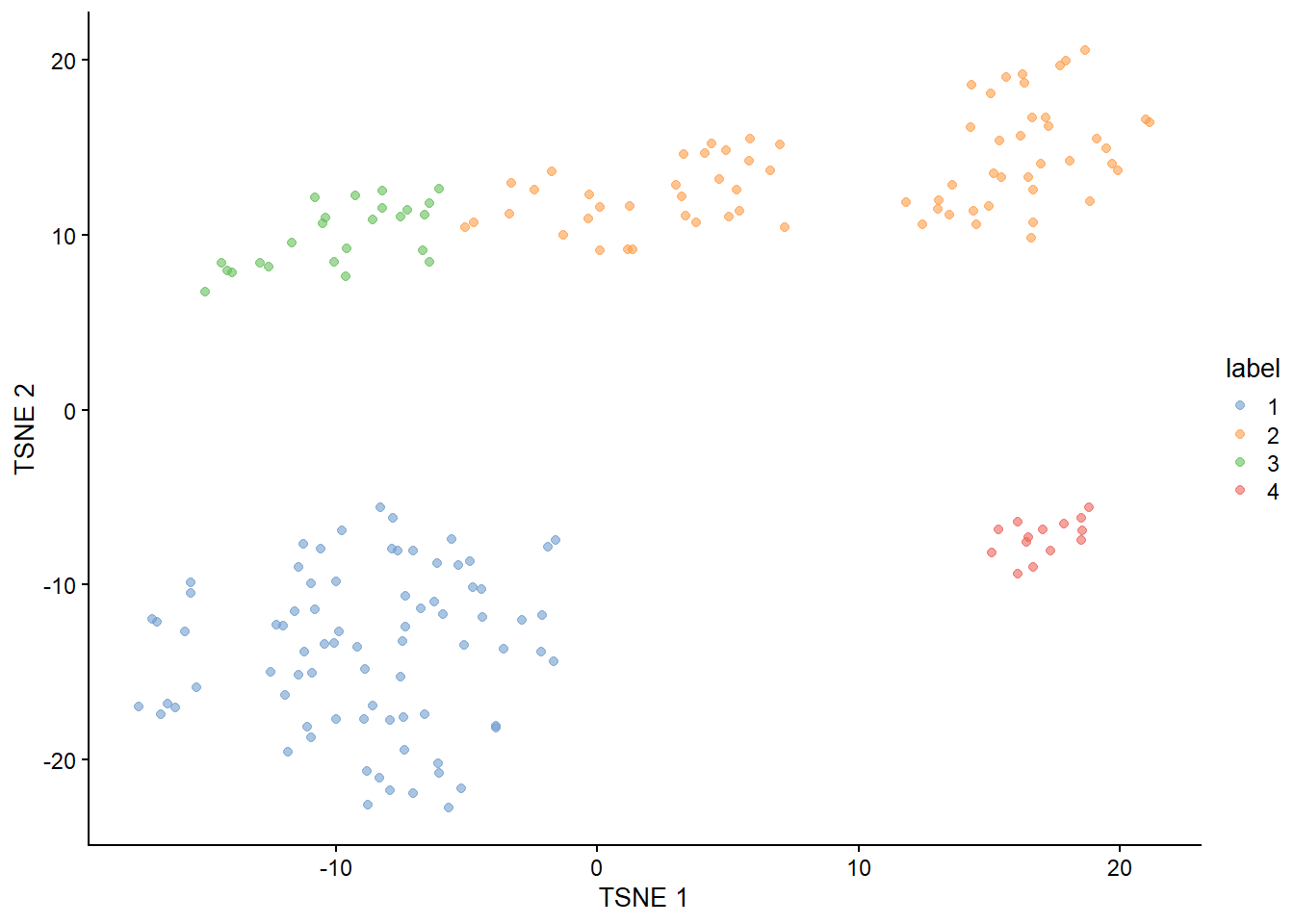
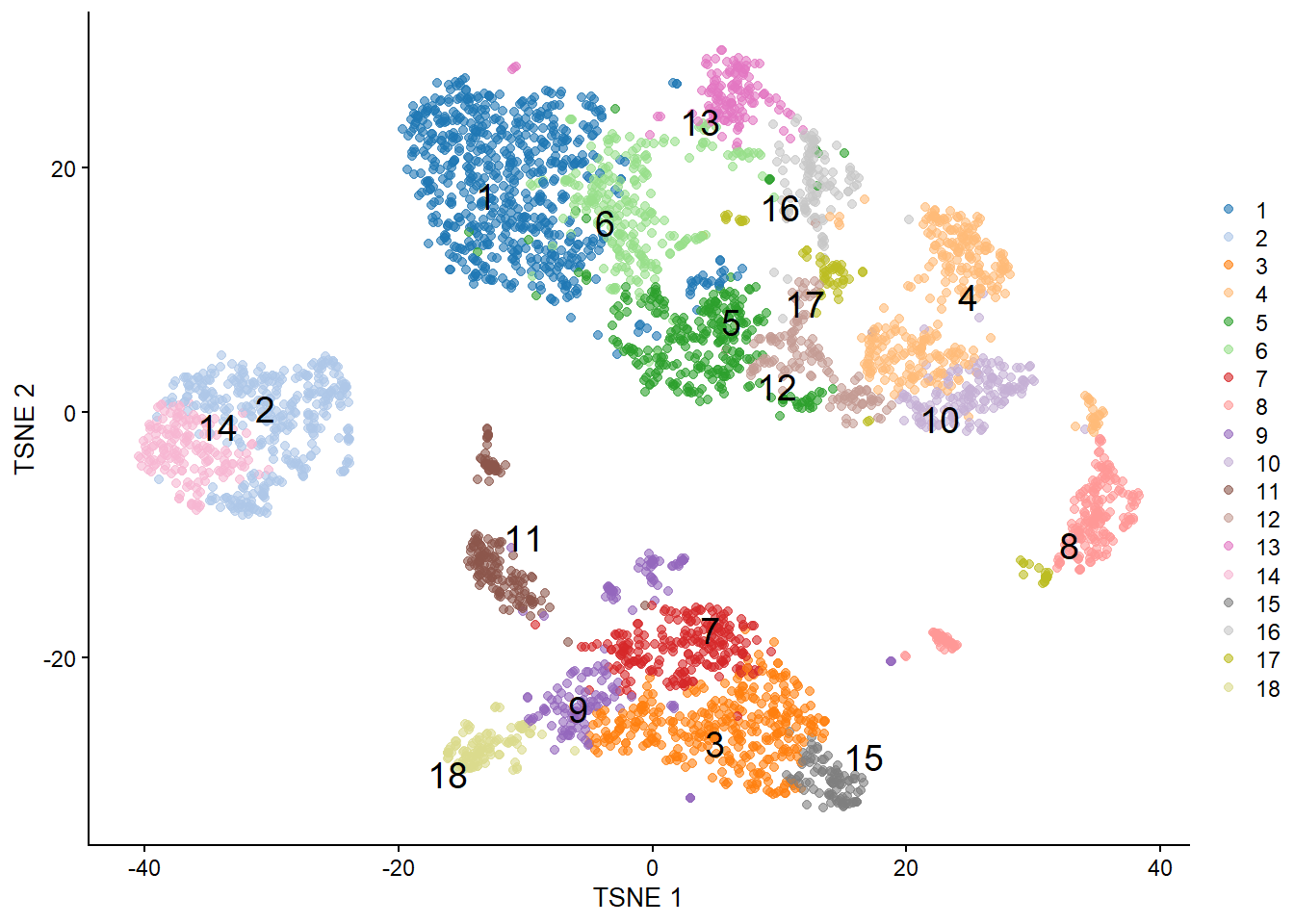
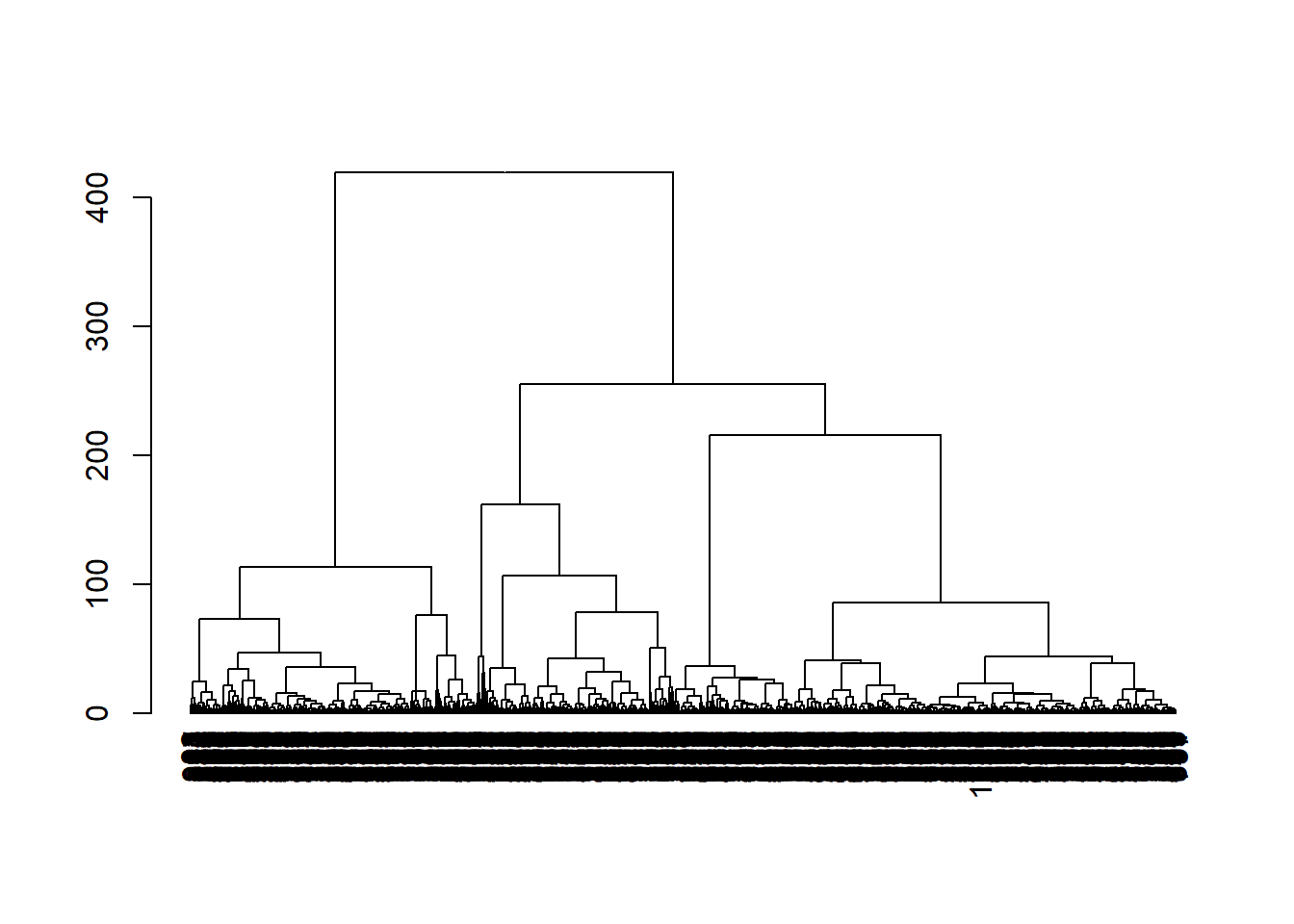
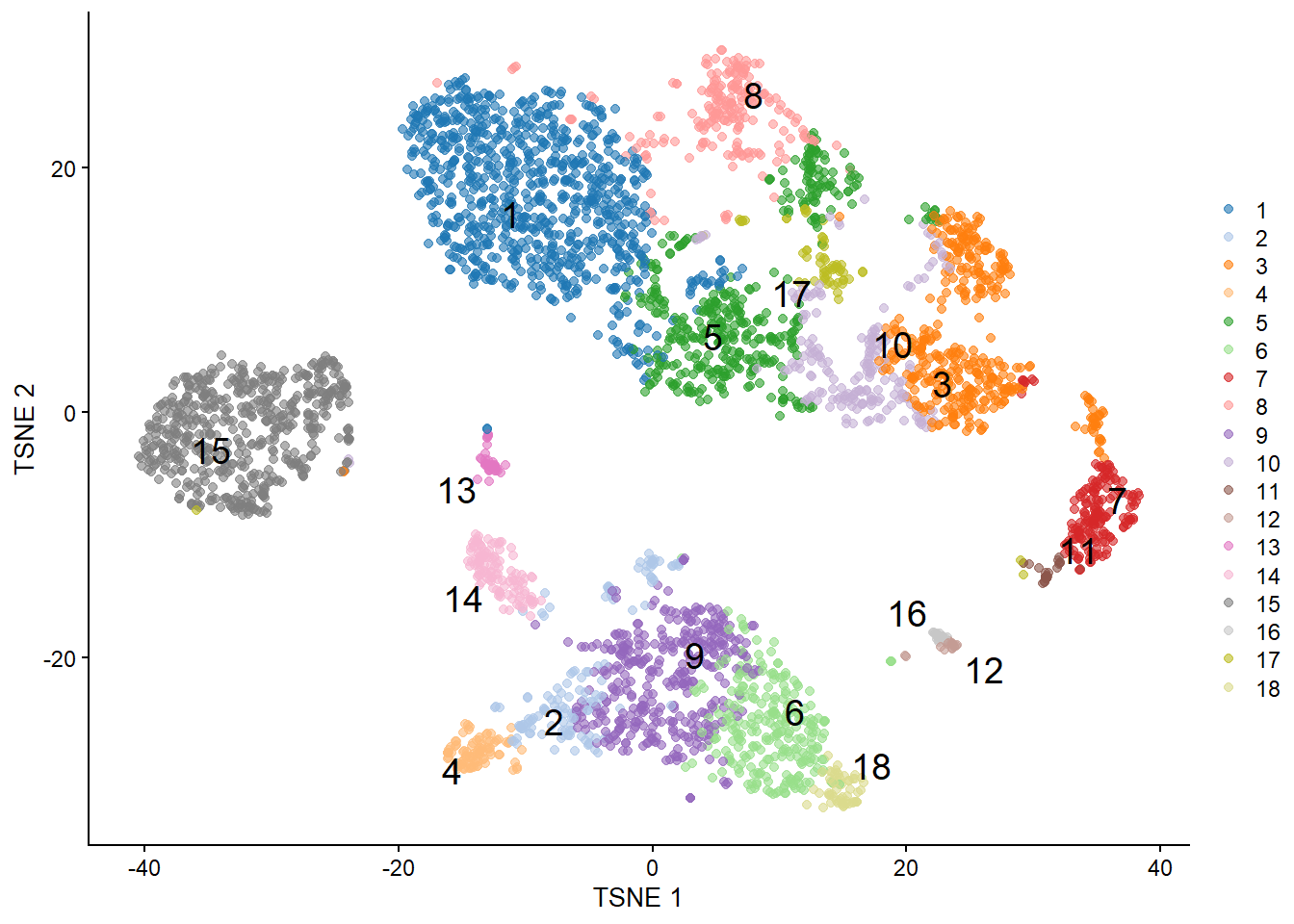
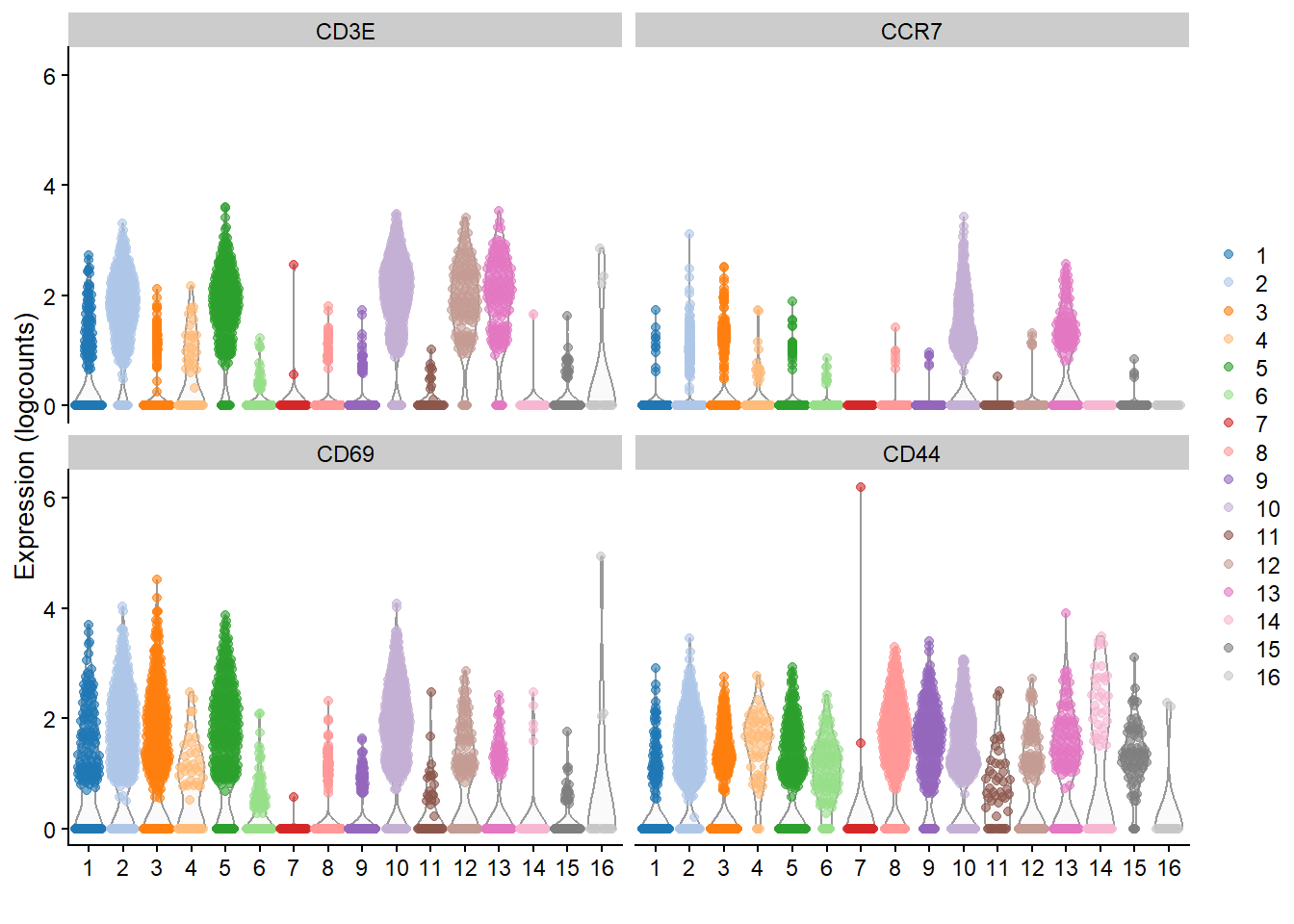
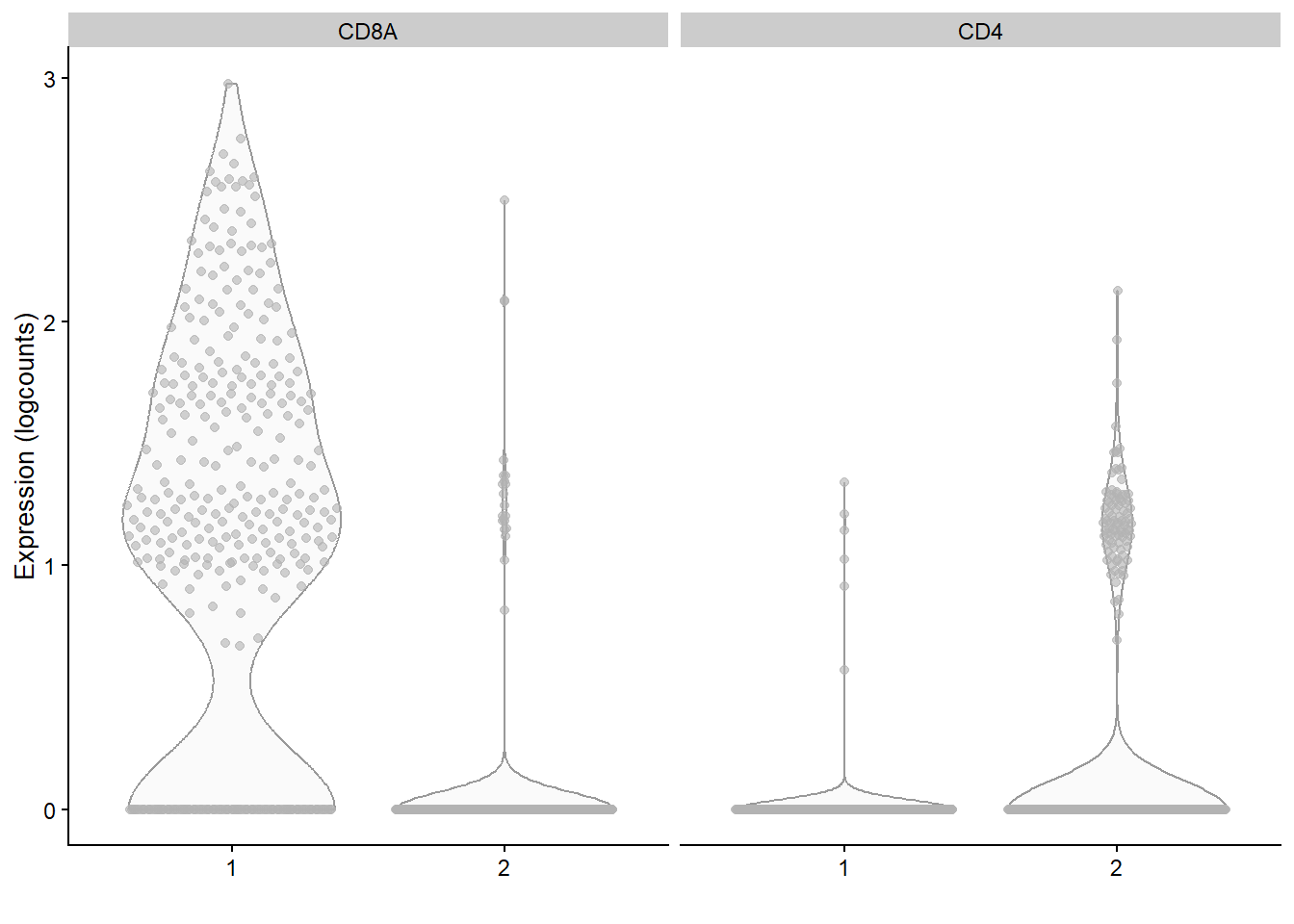
```{r} #| include: false conflicts_prefer(GenomicRanges::setdiff) #conflicts_prefer(dplyr::filter) ``` # 聚类 ```{r} #| label: loading-sce.pbmc_to_降维 #--- loading ---# library (DropletUtils)<- DropletUtils:: read10xCounts ("data/OSCA/raw_gene_bc_matrices/GRCh38" ,col.names = TRUE )#--- gene-annotation ---# library (scater)rownames (sce.pbmc) <- uniquifyFeatureNames (rowData (sce.pbmc)$ ID, rowData (sce.pbmc)$ Symbol)library (EnsDb.Hsapiens.v86)<- mapIds (EnsDb.Hsapiens.v86, keys= rowData (sce.pbmc)$ ID, column= "SEQNAME" , keytype= "GENEID" )#--- cell-detection ---# set.seed (100 )<- emptyDrops (counts (sce.pbmc))<- sce.pbmc[,which (e.out$ FDR <= 0.001 )]#--- quality-control ---# <- perCellQCMetrics (sce.pbmc, subsets= list (Mito= which (location== "MT" )))<- isOutlier (stats$ subsets_Mito_percent, type= "higher" )<- sce.pbmc[,! high.mito]#--- normalization ---# library (scran)set.seed (1000 )<- quickCluster (sce.pbmc)<- computeSumFactors (sce.pbmc, cluster= clusters)<- logNormCounts (sce.pbmc)#--- variance-modelling ---# set.seed (1001 )<- modelGeneVarByPoisson (sce.pbmc)<- getTopHVGs (dec.pbmc, prop= 0.1 )#--- dimensionality-reduction ---# set.seed (10000 )<- denoisePCA (sce.pbmc, subset.row= top.pbmc, technical= dec.pbmc)set.seed (100000 )<- runTSNE (sce.pbmc, dimred= "PCA" )set.seed (1000000 )<- runUMAP (sce.pbmc, dimred= "PCA" )``` ```{r} ``` ## Graph-based clustering ```{r} library (scran)<- clusterCells (sce.pbmc, use.dimred= "PCA" )table (nn.clusters)``` ```{r} library (scater)colLabels (sce.pbmc) <- nn.clustersplotReducedDim (sce.pbmc, "TSNE" , colour_by= "label" )``` ` walktrap` ```{r} library (bluster)<- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= SNNGraphParam (k= 10 , type= "rank" , cluster.fun= "walktrap" ))table (nn.clusters2)``` `clusterCells()` 中指定`full=TRUE` 来获取图本身,这样做将返回聚类分析期间使用的所有中间结构,包括 [ igraph ](https://cran.r-project.org/web/packages/igraph/index.html) 包中的图形对象。 该图可以使用力导向布局(force-directed layout)进行可视化```{r} <- clusterCells (sce.pbmc, use.dimred= "PCA" , full= TRUE )$ objects$ graph``` ```{r} #| label: fig-Force-directed-layout--shared-nearest-neighbor-graph set.seed (11000 )reducedDim (sce.pbmc, "force" ) <- igraph:: layout_with_fr (nn.clust.info$ objects$ graph)plotReducedDim (sce.pbmc, colour_by= "label" , dimred= "force" )``` ### 参数 #### ` k ` 最近邻数目 ```{r} # More resolved. .5 <- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (k= 5 ))table (clust.5 )``` ```{r} # Less resolved. .50 <- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (k= 50 ))table (clust.50 )``` #### ` type ` 边缘加权方法 `type="number" ` 将根据两个细胞之间共享的最近邻的数量对边进行加权。```{r} <- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (type= "number" ))table (clust.num)``` `type="jaccard" ` 将根据两组最近邻的 Jaccard 指数对边缘进行加权。```{r} <- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (type= "jaccard" ))table (clust.jaccard)``` ```{r} <- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= KNNGraphParam ())table (clust.none)``` #### ` cluster.fun ` 簇识别方法 [ igraph ](https://cran.r-project.org/web/packages/igraph/index.html) ```{r} library (igraph)<- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (cluster.fun= "walktrap" ))<- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (cluster.fun= "louvain" ))<- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (cluster.fun= "infomap" ))<- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (cluster.fun= "fast_greedy" ))<- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (cluster.fun= "label_prop" ))<- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= NNGraphParam (cluster.fun= "leading_eigen" ))``` ```{r} #| label: fig-Infomap_vs_Walktrap library (pheatmap)# Using a large pseudo-count for a smoother color transition # between 0 and 1 cell in each 'tab'. <- table (paste ("Infomap" , clust.infomap), paste ("Walktrap" , clust.walktrap))<- pheatmap (log10 (tab+ 10 ), main= "Infomap vs Walktrap" ,color= viridis:: viridis (100 ), silent= TRUE ):: grid.arrange (ivw[[4 ]])``` ```{r} #| label: fig-Fast-greedy_vs_Walktrap <- table (paste ("Fast" , clust.fast), paste ("Walktrap" , clust.walktrap))<- pheatmap (log10 (tab+ 10 ), main= "Fast-greedy vs Walktrap" ,color= viridis:: viridis (100 ), silent= TRUE ):: grid.arrange (fvw[[4 ]])``` [ scran ](https://bioconductor.org/packages/3.18/bioc/html/scran.html) 的管道默认为基于秩(rank)的权重,后跟 Walktrap 聚类。 相比之下,Seurat 使用基于 Jaccard 的权重,然后是 Louvain 聚类。 ## K-means 聚类 ```{r} set.seed (100 )<- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= KmeansParam (centers= 10 ))table (clust.kmeans)``` ```{r} colLabels (sce.pbmc) <- clust.kmeansplotReducedDim (sce.pbmc, "TSNE" , colour_by= "label" )``` ```{r} set.seed (100 )<- clusterCells (sce.pbmc, use.dimred= "PCA" , BLUSPARAM= KmeansParam (centers= 20 ))table (clust.kmeans2)colLabels (sce.pbmc) <- clust.kmeans2plotTSNE (sce.pbmc, colour_by= "label" , text_by= "label" )``` ### mini-batch k-means [ mbkmeans ](https://bioconductor.org/packages/3.18/bioc/html/mbkmeans.html) ```{r} set.seed (100 )<- clusterCells (sce.pbmc, use.dimred= "PCA" ,BLUSPARAM= MbkmeansParam (centers= 10 ))table (clust.mbkmeans)``` 1. 它隐式偏向于半径相等的球形簇,这可能会导致在包含大小和形状不规则的分组的数据集上出现不直观的分区。2. 簇个数(质心数,centers) k 必须事先指定,并表示聚类分辨率的上限。k小于细胞类型数量时将始终导致两3. 它取决于随机选择的初始质心。 这需要多次运行来验证簇是否稳定。### "two-step" mode:Kmeans-nnGraph ```{r} #| label: fig-K-Graph_cluster # Setting the seed due to the randomness of k-means. set.seed (0101010 )<- clusterCells (sce.pbmc, use.dimred= "PCA" ,BLUSPARAM= TwoStepParam (first= KmeansParam (centers= 1000 ),second= NNGraphParam (k= 5 )table (kgraph.clusters)plotTSNE (sce.pbmc, colour_by= I (kgraph.clusters))``` ## Hierarchical clustering ```{r} #--- loading ---# library (scRNAseq).416 b <- LunSpikeInData (which= "416b" ) .416 b$ block <- factor (sce.416 b$ block)#--- gene-annotation ---# library (AnnotationHub)<- AnnotationHub ()[["AH73905" ]]rowData (sce.416 b)$ ENSEMBL <- rownames (sce.416 b)rowData (sce.416 b)$ SYMBOL <- mapIds (ens.mm.v97, keys= rownames (sce.416 b),keytype= "GENEID" , column= "SYMBOL" )rowData (sce.416 b)$ SEQNAME <- mapIds (ens.mm.v97, keys= rownames (sce.416 b),keytype= "GENEID" , column= "SEQNAME" )library (scater)rownames (sce.416 b) <- uniquifyFeatureNames (rowData (sce.416 b)$ ENSEMBL, rowData (sce.416 b)$ SYMBOL)#--- quality-control ---# <- which (rowData (sce.416 b)$ SEQNAME== "MT" )<- perCellQCMetrics (sce.416 b, subsets= list (Mt= mito))<- quickPerCellQC (stats, percent_subsets= c ("subsets_Mt_percent" ,"altexps_ERCC_percent" ), batch= sce.416 b$ block).416 b <- sce.416 b[,! qc$ discard]#--- normalization ---# library (scran).416 b <- computeSumFactors (sce.416 b).416 b <- logNormCounts (sce.416 b)#--- variance-modelling ---# .416 b <- modelGeneVarWithSpikes (sce.416 b, "ERCC" , block= sce.416 b$ block)<- getTopHVGs (dec.416 b, prop= 0.1 )#--- batch-correction ---# library (limma)assay (sce.416 b, "corrected" ) <- removeBatchEffect (logcounts (sce.416 b), design= model.matrix (~ sce.416 b$ phenotype), batch= sce.416 b$ block)#--- dimensionality-reduction ---# .416 b <- runPCA (sce.416 b, ncomponents= 10 , subset_row= chosen.hvgs,exprs_values= "corrected" , BSPARAM= BiocSingular:: ExactParam ())set.seed (1010 ).416 b <- runTSNE (sce.416 b, dimred= "PCA" , perplexity= 10 )``` ```{r} .416 b``` ```{r} #| label: fig-hclust #| fig-cap: "根据致癌基因诱导状态(红色为诱导,蓝色为对照)和起始板(浅色或深色)进行着色" .416 b <- clusterCells (sce.416 b, use.dimred= "PCA" ,BLUSPARAM= HclustParam (method= "ward.D2" ), full= TRUE ).416 b <- hclust.416 b$ objects$ hclust# Making a prettier dendrogram. library (dendextend).416 b$ labels <- seq_along (tree.416 b$ labels)<- as.dendrogram (tree.416 b, hang= 0.1 )<- paste0 (sce.416 b$ block, "." , sub (" .*" , "" , sce.416 b$ phenotype))labels_colors (dend) <- c ("20160113.wild" = "blue" ,"20160113.induced" = "red" ,"20160325.wild" = "dodgerblue" ,"20160325.induced" = "salmon" order.dendrogram (dend)]plot (dend)``` [ dynamicTreeCut ](https://cran.r-project.org/web/packages/dynamicTreeCut/index.html) ```{r} #| label: fig-hclust-dynamicTreeCut #| fig-cap: "根据dynamic Cut中分配的聚类标识进行着色" <- clusterCells (sce.416 b, use.dimred= "PCA" ,BLUSPARAM= HclustParam (method= "ward.D2" , cut.dynamic= TRUE ,cut.params= list (minClusterSize= 10 , deepSplit= 1 )))table (hclust.dyn)labels_colors (dend) <- as.integer (hclust.dyn)[order.dendrogram (dend)]plot (dend)``` ```{r} #| label: fig-tsne-hclust-dynamicTreeCut colLabels (sce.416 b) <- factor (hclust.dyn)plotReducedDim (sce.416 b, "TSNE" , colour_by= "label" )``` ### "two-step" mode :Kmeans-Hierarchical ```{r} #| label: fig-K-H_cluster #| fig-cap: "根据k-means/hierarchical聚类组合中分配的集群的身份进行着色 " # Setting the seed due to the randomness of k-means. set.seed (1111 )<- clusterCells (sce.pbmc, use.dimred= "PCA" ,BLUSPARAM= TwoStepParam (first= KmeansParam (centers= 1000 ),second= HclustParam (method= "ward.D2" , cut.dynamic= TRUE ,cut.param= list (deepSplit= 3 )) # for higher resolution. full= TRUE table (khclust.info$ clusters)plotTSNE (sce.pbmc, colour_by= I (khclust.info$ clusters), text_by= I (khclust.info$ clusters))``` ```{r} #| label: fig-dendrogram-centroids <- khclust.info$ objects$ first<- khclust.info$ objects$ second$ hclust<- match (as.integer (tree.pbmc$ labels), k.stats$ cluster)<- khclust.info$ clusters[m]# TODO : expose scater color palette for easier re-use, # given that the default colors start getting recycled. <- as.dendrogram (tree.pbmc, hang= 0.1 )labels_colors (dend) <- as.integer (final.clusters)[order.dendrogram (dend)]plot (dend)``` ### "two-step" mode :Kmeans-Affinity `q=` 控制,该参数定义了样本将自己视为示例并因此形成自己的簇的强度。```{r} #| label: fig-Kmeans-Affinity # Setting the seed due to the randomness of k-means. library (apcluster)set.seed (1111 )<- clusterCells (sce.pbmc, use.dimred= "PCA" ,BLUSPARAM= TwoStepParam (first= KmeansParam (centers= 1000 ),second= AffinityParam (q= 0.1 ) # larger q => more clusters full= TRUE table (kaclust.info$ clusters)plotTSNE (sce.pbmc, colour_by= I (kaclust.info$ clusters), text_by= I (kaclust.info$ clusters))``` ## Subclustering ```{r} <- clusterCells (sce.pbmc, use.dimred= "PCA" )plotExpression (sce.pbmc, features= c ("CD3E" , "CCR7" , "CD69" , "CD44" ),x= I (clust.full), colour_by= I (clust.full))``` ```{r} # Repeating modelling and PCA on the subset. <- 10 L<- sce.pbmc[,clust.full== memory]<- modelGeneVar (sce.memory)<- denoisePCA (sce.memory, technical= dec.memory,subset.row= getTopHVGs (dec.memory, n= 5000 ))``` ```{r} <- buildSNNGraph (sce.memory, use.dimred= "PCA" )<- igraph:: cluster_walktrap (g.memory)$ membershipplotExpression (sce.memory, features= c ("CD8A" , "CD4" ),x= I (factor (clust.memory)))``` `quickSubCluster()` ```{r} set.seed (1000010 )<- quickSubCluster (sce.pbmc, groups= clust.full,prepFUN= function (x) { # Preparing the subsetted SCE for clustering. <- modelGeneVar (x)<- denoisePCA (x, technical= dec,subset.row= getTopHVGs (dec, prop= 0.1 ),BSPARAM= BiocSingular:: IrlbaParam ())clusterFUN= function (x) { # Performing the subclustering in the subset. <- buildSNNGraph (x, use.dimred= "PCA" , k= 20 ):: cluster_walktrap (g)$ membership# One SingleCellExperiment object per parent cluster: names (subcluster.out)# Looking at the subclustering for one example: table (subcluster.out[[1 ]]$ subcluster)```