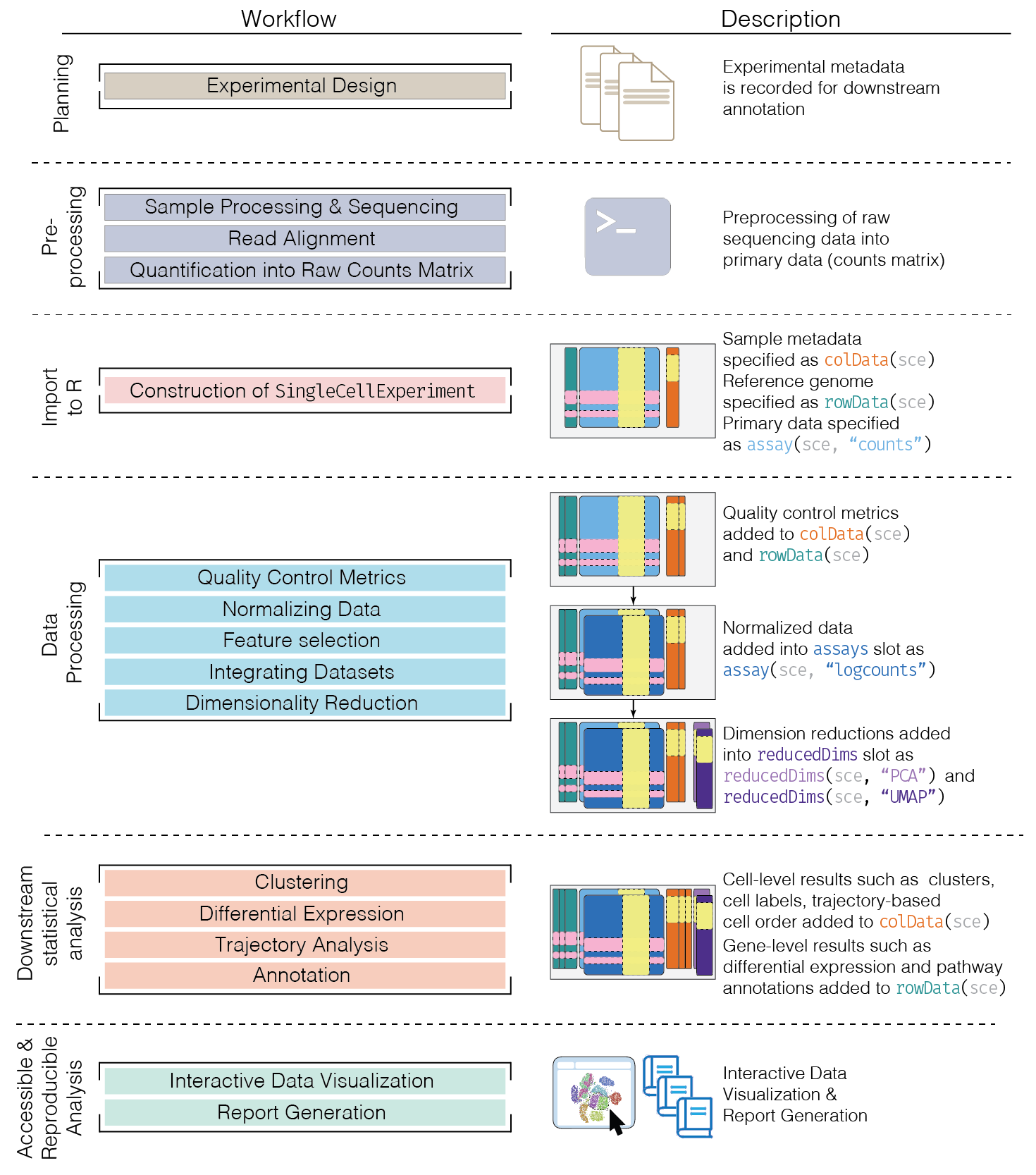
典型scRNA-seq分析工作流如 Figure fig-workflow 所示。
表达矩阵
质量控制
表达值标准化
特征选择
降维
聚类
标记基因检测
细胞类型注释
差异表达分析
轨迹分析
快速开始(简单)
Code
sce <- scRNAseq::MacoskoRetinaData()
sce
#> class: SingleCellExperiment
#> dim: 24658 49300
#> metadata(0):
#> assays(1): counts
#> rownames(24658): KITL TMTC3 ... 1110059M19RIK GM20861
#> rowData names(0):
#> colnames(49300): r1_GGCCGCAGTCCG r1_CTTGTGCGGGAA ... p1_TAACGCGCTCCT
#> p1_ATTCTTGTTCTT
#> colData names(2): cell.id cluster
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
Code
counts <- assay(sce, "counts")
colData(sce)
#> DataFrame with 49300 rows and 2 columns
#> cell.id cluster
#> <character> <integer>
#> r1_GGCCGCAGTCCG r1_GGCCGCAGTCCG 2
#> r1_CTTGTGCGGGAA r1_CTTGTGCGGGAA 2
#> r1_GCGCAACTGCTC r1_GCGCAACTGCTC 2
#> r1_GATTGGGAGGCA r1_GATTGGGAGGCA 2
#> r1_CCTCCTAGTTGG r1_CCTCCTAGTTGG NA
#> ... ... ...
#> p1_TCAAAAGCCGGG p1_TCAAAAGCCGGG 24
#> p1_ATTAAGTTCCAA p1_ATTAAGTTCCAA 34
#> p1_CTGTCTGAGACC p1_CTGTCTGAGACC 2
#> p1_TAACGCGCTCCT p1_TAACGCGCTCCT 24
#> p1_ATTCTTGTTCTT p1_ATTCTTGTTCTT 24
# Quality control (using mitochondrial genes)
is.mito <- str_detect(rownames(sce),"^MT-") # 线粒体基因
sum(is.mito) # 31个
#> [1] 31
library(scater)
library(scuttle)
#?perCellQCMetrics
scuttle::perCellQCMetrics(sce)
#> DataFrame with 49300 rows and 3 columns
#> sum detected total
#> <numeric> <integer> <numeric>
#> r1_GGCCGCAGTCCG 37487 7243 37487
#> r1_CTTGTGCGGGAA 32047 6933 32047
#> r1_GCGCAACTGCTC 28148 6397 28148
#> r1_GATTGGGAGGCA 20367 5740 20367
#> r1_CCTCCTAGTTGG 19560 5779 19560
#> ... ... ... ...
#> p1_TCAAAAGCCGGG 817 537 817
#> p1_ATTAAGTTCCAA 817 574 817
#> p1_CTGTCTGAGACC 817 637 817
#> p1_TAACGCGCTCCT 816 488 816
#> p1_ATTCTTGTTCTT 816 484 816
qcstats <- perCellQCMetrics(sce, subsets=list(Mito=is.mito))
qcstats
#> DataFrame with 49300 rows and 6 columns
#> sum detected subsets_Mito_sum subsets_Mito_detected
#> <numeric> <integer> <numeric> <integer>
#> r1_GGCCGCAGTCCG 37487 7243 427 14
#> r1_CTTGTGCGGGAA 32047 6933 503 15
#> r1_GCGCAACTGCTC 28148 6397 460 13
#> r1_GATTGGGAGGCA 20367 5740 326 11
#> r1_CCTCCTAGTTGG 19560 5779 264 9
#> ... ... ... ... ...
#> p1_TCAAAAGCCGGG 817 537 13 4
#> p1_ATTAAGTTCCAA 817 574 10 5
#> p1_CTGTCTGAGACC 817 637 24 7
#> p1_TAACGCGCTCCT 816 488 27 5
#> p1_ATTCTTGTTCTT 816 484 16 4
#> subsets_Mito_percent total
#> <numeric> <numeric>
#> r1_GGCCGCAGTCCG 1.13906 37487
#> r1_CTTGTGCGGGAA 1.56957 32047
#> r1_GCGCAACTGCTC 1.63422 28148
#> r1_GATTGGGAGGCA 1.60063 20367
#> r1_CCTCCTAGTTGG 1.34969 19560
#> ... ... ...
#> p1_TCAAAAGCCGGG 1.59119 817
#> p1_ATTAAGTTCCAA 1.22399 817
#> p1_CTGTCTGAGACC 2.93758 817
#> p1_TAACGCGCTCCT 3.30882 816
#> p1_ATTCTTGTTCTT 1.96078 816
#?quickPerCellQC
scuttle::quickPerCellQC(sce) |> colData()
#> DataFrame with 49287 rows and 8 columns
#> cell.id cluster sum detected total
#> <character> <integer> <numeric> <integer> <numeric>
#> r1_GGCCGCAGTCCG r1_GGCCGCAGTCCG 2 37487 7243 37487
#> r1_CTTGTGCGGGAA r1_CTTGTGCGGGAA 2 32047 6933 32047
#> r1_GCGCAACTGCTC r1_GCGCAACTGCTC 2 28148 6397 28148
#> r1_GATTGGGAGGCA r1_GATTGGGAGGCA 2 20367 5740 20367
#> r1_CCTCCTAGTTGG r1_CCTCCTAGTTGG NA 19560 5779 19560
#> ... ... ... ... ... ...
#> p1_TCAAAAGCCGGG p1_TCAAAAGCCGGG 24 817 537 817
#> p1_ATTAAGTTCCAA p1_ATTAAGTTCCAA 34 817 574 817
#> p1_CTGTCTGAGACC p1_CTGTCTGAGACC 2 817 637 817
#> p1_TAACGCGCTCCT p1_TAACGCGCTCCT 24 816 488 816
#> p1_ATTCTTGTTCTT p1_ATTCTTGTTCTT 24 816 484 816
#> low_lib_size low_n_features discard
#> <outlier.filter> <outlier.filter> <logical>
#> r1_GGCCGCAGTCCG FALSE FALSE FALSE
#> r1_CTTGTGCGGGAA FALSE FALSE FALSE
#> r1_GCGCAACTGCTC FALSE FALSE FALSE
#> r1_GATTGGGAGGCA FALSE FALSE FALSE
#> r1_CCTCCTAGTTGG FALSE FALSE FALSE
#> ... ... ... ...
#> p1_TCAAAAGCCGGG FALSE FALSE FALSE
#> p1_ATTAAGTTCCAA FALSE FALSE FALSE
#> p1_CTGTCTGAGACC FALSE FALSE FALSE
#> p1_TAACGCGCTCCT FALSE FALSE FALSE
#> p1_ATTCTTGTTCTT FALSE FALSE FALSE
quickPerCellQC(qcstats)
#> DataFrame with 49300 rows and 3 columns
#> low_lib_size low_n_features discard
#> <outlier.filter> <outlier.filter> <logical>
#> 1 FALSE FALSE FALSE
#> 2 FALSE FALSE FALSE
#> 3 FALSE FALSE FALSE
#> 4 FALSE FALSE FALSE
#> 5 FALSE FALSE FALSE
#> ... ... ... ...
#> 49296 FALSE FALSE FALSE
#> 49297 FALSE FALSE FALSE
#> 49298 FALSE FALSE FALSE
#> 49299 FALSE FALSE FALSE
#> 49300 FALSE FALSE FALSE
filtered <- quickPerCellQC(qcstats, percent_subsets="subsets_Mito_percent")
filtered
#> DataFrame with 49300 rows and 4 columns
#> low_lib_size low_n_features high_subsets_Mito_percent discard
#> <outlier.filter> <outlier.filter> <outlier.filter> <logical>
#> 1 FALSE FALSE FALSE FALSE
#> 2 FALSE FALSE FALSE FALSE
#> 3 FALSE FALSE FALSE FALSE
#> 4 FALSE FALSE FALSE FALSE
#> 5 FALSE FALSE FALSE FALSE
#> ... ... ... ... ...
#> 49296 FALSE FALSE FALSE FALSE
#> 49297 FALSE FALSE FALSE FALSE
#> 49298 FALSE FALSE FALSE FALSE
#> 49299 FALSE FALSE FALSE FALSE
#> 49300 FALSE FALSE FALSE FALSE
sce <- sce[, !filtered$discard]
sce
#> class: SingleCellExperiment
#> dim: 24658 45877
#> metadata(0):
#> assays(1): counts
#> rownames(24658): KITL TMTC3 ... 1110059M19RIK GM20861
#> rowData names(0):
#> colnames(45877): r1_GGCCGCAGTCCG r1_CTTGTGCGGGAA ... p1_TAACGCGCTCCT
#> p1_ATTCTTGTTCTT
#> colData names(2): cell.id cluster
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
Code
# Normalization
sce <- logNormCounts(sce)
assays(sce)
#> List of length 2
#> names(2): counts logcounts
Code
# Feature selection
library(scran)
dec <- scran::modelGeneVar(sce) # ?modelGeneVar 对每个基因的对数表达谱的方差进行建模,根据拟合的均值-方差趋势将其分解为技术差异和生物学差异。
dec
#> DataFrame with 24658 rows and 6 columns
#> mean total tech bio p.value
#> <numeric> <numeric> <numeric> <numeric> <numeric>
#> KITL 0.00884872 0.011881960 0.011546899 3.35060e-04 0.4118020
#> TMTC3 0.05143737 0.075214907 0.066708042 8.50687e-03 0.1636316
#> CEP290 0.57977970 0.841422026 0.689765311 1.51657e-01 0.0456074
#> 4930430F08RIK 0.05284121 0.069214001 0.068514734 6.99267e-04 0.4687536
#> 1700017N19RIK 0.00047060 0.000537273 0.000614846 -7.75727e-05 0.8337798
#> ... ... ... ... ... ...
#> VSIG1 7.47516e-06 2.56352e-06 9.76708e-06 -7.20357e-06 1
#> GM16390 0.00000e+00 0.00000e+00 0.00000e+00 0.00000e+00 NaN
#> GM25207 3.60098e-06 5.94891e-07 4.70506e-06 -4.11017e-06 1
#> 1110059M19RIK 0.00000e+00 0.00000e+00 0.00000e+00 0.00000e+00 NaN
#> GM20861 0.00000e+00 0.00000e+00 0.00000e+00 0.00000e+00 NaN
#> FDR
#> <numeric>
#> KITL 1.000000
#> TMTC3 1.000000
#> CEP290 0.791672
#> 4930430F08RIK 1.000000
#> 1700017N19RIK 1.000000
#> ... ...
#> VSIG1 1
#> GM16390 NaN
#> GM25207 1
#> 1110059M19RIK NaN
#> GM20861 NaN
hvg <- getTopHVGs(dec, prop=0.1) # ?getTopHVGs 根据 modelGeneVar() 或相关函数的方差建模统计数据,定义一组高度可变的基因。
length(hvg)
#> [1] 914
Code
# Visualization
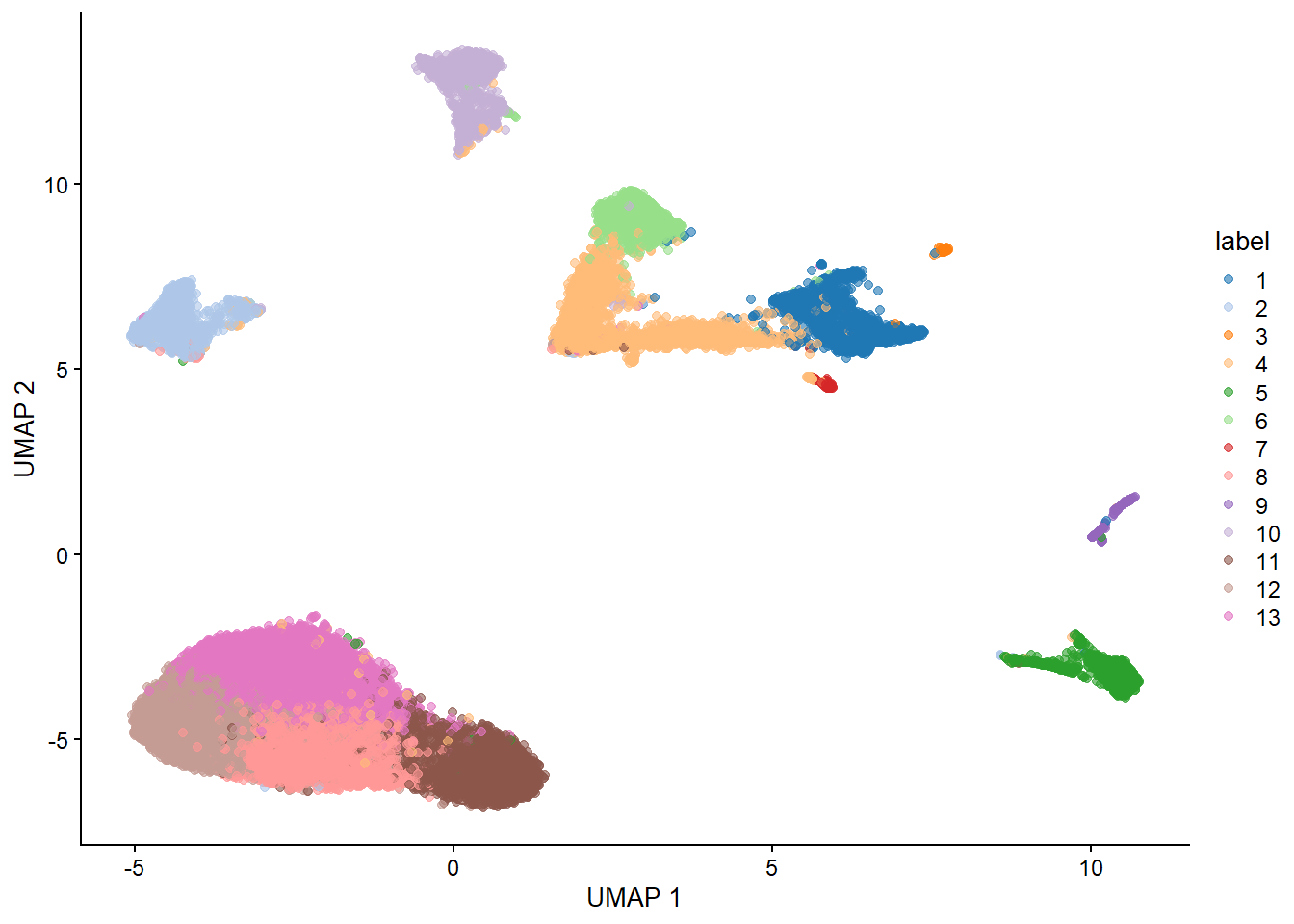
sce <- runUMAP(sce, dimred = 'PCA')
plotUMAP(sce, colour_by="label")
Code
# Marker detection
markers <- findMarkers(sce, test.type="wilcox", direction="up", lfc=1)# ?findMarkers 通过检测细胞组配对之间的差异表达,找到细胞组(如聚类)的候选标记基因。
快速开@fig-Malat1始(多批次)
Code
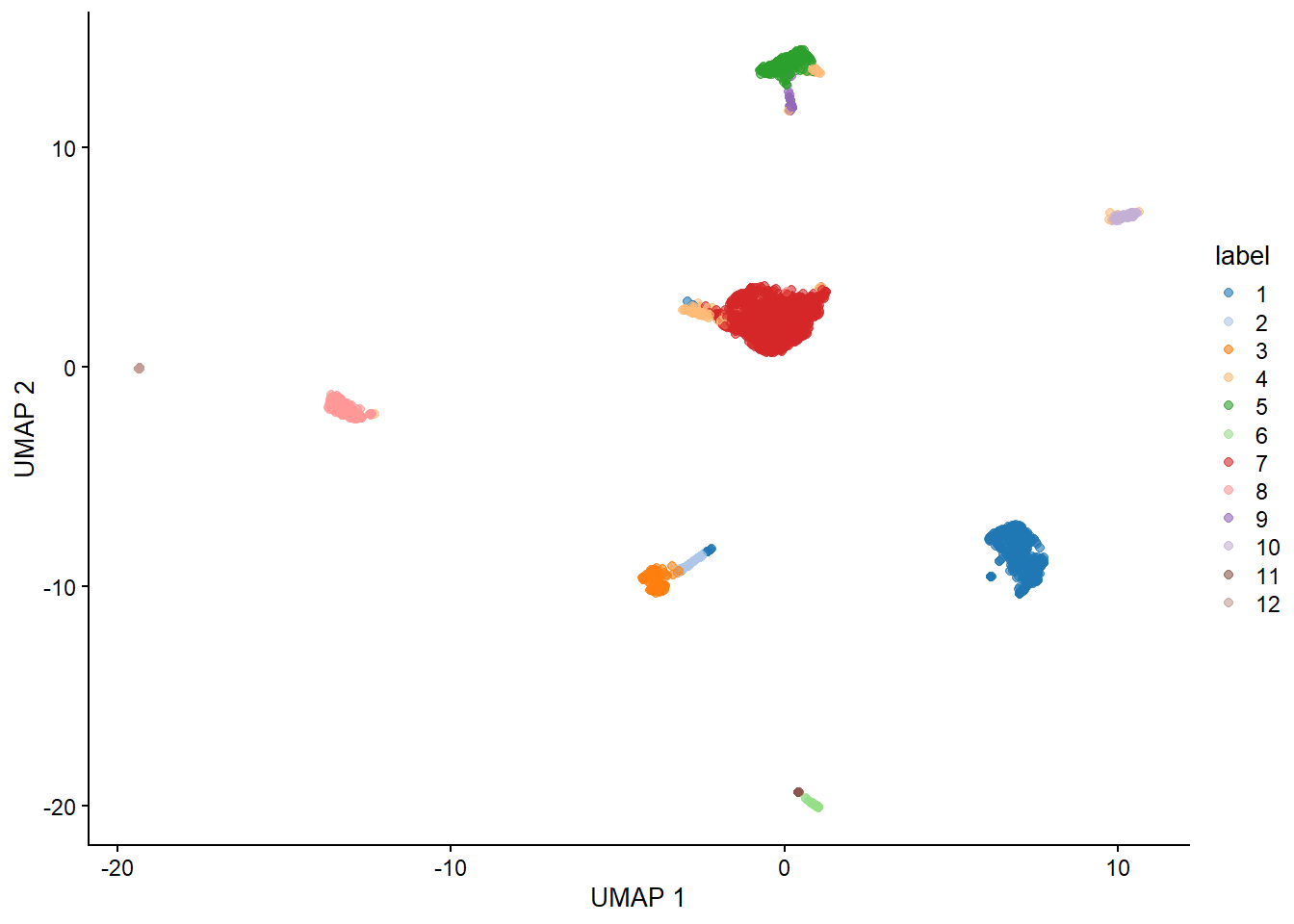
sce1 <- runUMAP(sce1, dimred = 'corrected')
plotUMAP(sce1, colour_by="label")
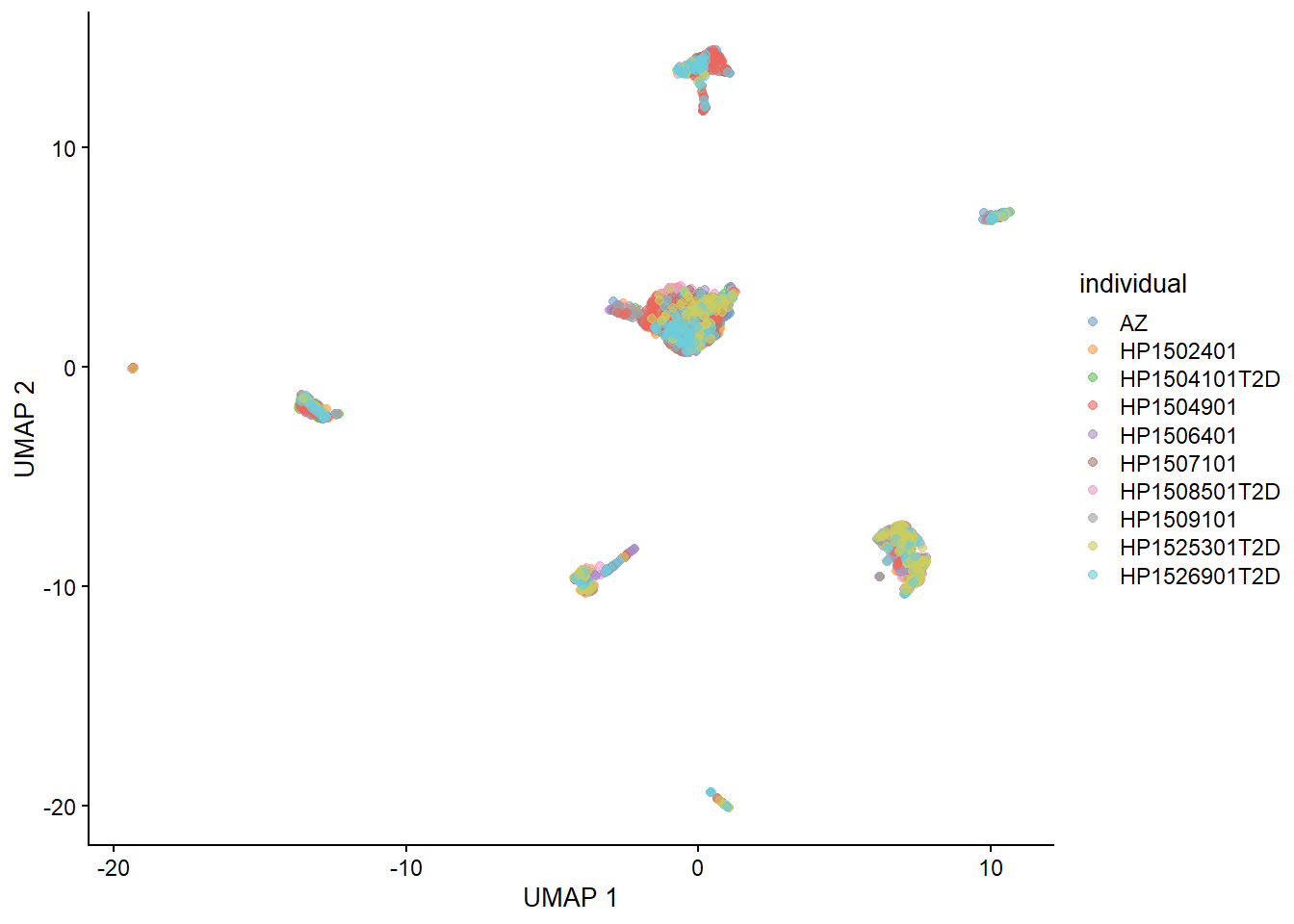
plotUMAP(sce1, colour_by="individual")
Figure 5.3: 胰腺数据集的 UMAP 图,其中每个点都是一个细胞,并由分配的聚类标识(左)或起源个体(右)着色。
Code
# Marker detection, blocking on the individual of origin.
markers1 <- findMarkers(sce1, test.type="wilcox", direction="up", lfc=1)