```{r}
#| include: false
conflicts_prefer(GenomicRanges::setdiff)
# conflicts_prefer(dplyr::filter)
```
# 标记基因检测
标记基因检测最直接的方法包括检测簇之间的差异表达。
```{r loading-sce.pbmc_to_clustering}
#--- loading ---#
library(DropletUtils)
sce.pbmc <- DropletUtils::read10xCounts("data/OSCA/raw_gene_bc_matrices/GRCh38",
col.names = TRUE)
#--- gene-annotation ---#
library(scater)
rownames(sce.pbmc) <- uniquifyFeatureNames(
rowData(sce.pbmc)$ID, rowData(sce.pbmc)$Symbol)
library(EnsDb.Hsapiens.v86)
location <- mapIds(EnsDb.Hsapiens.v86, keys=rowData(sce.pbmc)$ID,
column="SEQNAME", keytype="GENEID")
#--- cell-detection ---#
set.seed(100)
e.out <- emptyDrops(counts(sce.pbmc))
sce.pbmc <- sce.pbmc[,which(e.out$FDR <= 0.001)]
#--- quality-control ---#
stats <- perCellQCMetrics(sce.pbmc, subsets=list(Mito=which(location=="MT")))
high.mito <- isOutlier(stats$subsets_Mito_percent, type="higher")
sce.pbmc <- sce.pbmc[,!high.mito]
#--- normalization ---#
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.pbmc)
sce.pbmc <- computeSumFactors(sce.pbmc, cluster=clusters)
sce.pbmc <- logNormCounts(sce.pbmc)
#--- variance-modelling ---#
set.seed(1001)
dec.pbmc <- modelGeneVarByPoisson(sce.pbmc)
top.pbmc <- getTopHVGs(dec.pbmc, prop=0.1)
#--- dimensionality-reduction ---#
set.seed(10000)
sce.pbmc <- denoisePCA(sce.pbmc, subset.row=top.pbmc, technical=dec.pbmc)
set.seed(100000)
sce.pbmc <- runTSNE(sce.pbmc, dimred="PCA")
set.seed(1000000)
sce.pbmc <- runUMAP(sce.pbmc, dimred="PCA")
#--- clustering ---#
g <- buildSNNGraph(sce.pbmc, k=10, use.dimred = 'PCA')
clust <- igraph::cluster_walktrap(g)$membership
colLabels(sce.pbmc) <- factor(clust)
```
```{r}
sce.pbmc
```
## 通过成对比较对标记基因计分
`scoreMarkers()`
`self.average` 在X簇中的某基因平均对数表达值
`other.average` 所有其他簇的总体平均值
`self.detected` 在X簇中检测到某基因表达的细胞比例
`other.detected` 所有其他簇的平均检测到的细胞比例
`AUC` ,`logFC.cohen` ,`log.C.detected` 成对比较中生成的效应值汇总
AUC 或 Cohen's d 通常是通用标记检测的最佳选择,因为无论表达值的大小如何,它们都是有效的。 检测到表达值的细胞比例的对数倍数变化对于识别表达的binary changes特别有用。
```{r}
library(scran)
marker.info <- scoreMarkers(sce.pbmc, colLabels(sce.pbmc))
marker.info
colnames(marker.info[["1"]]) # statistics for cluster 1.
```
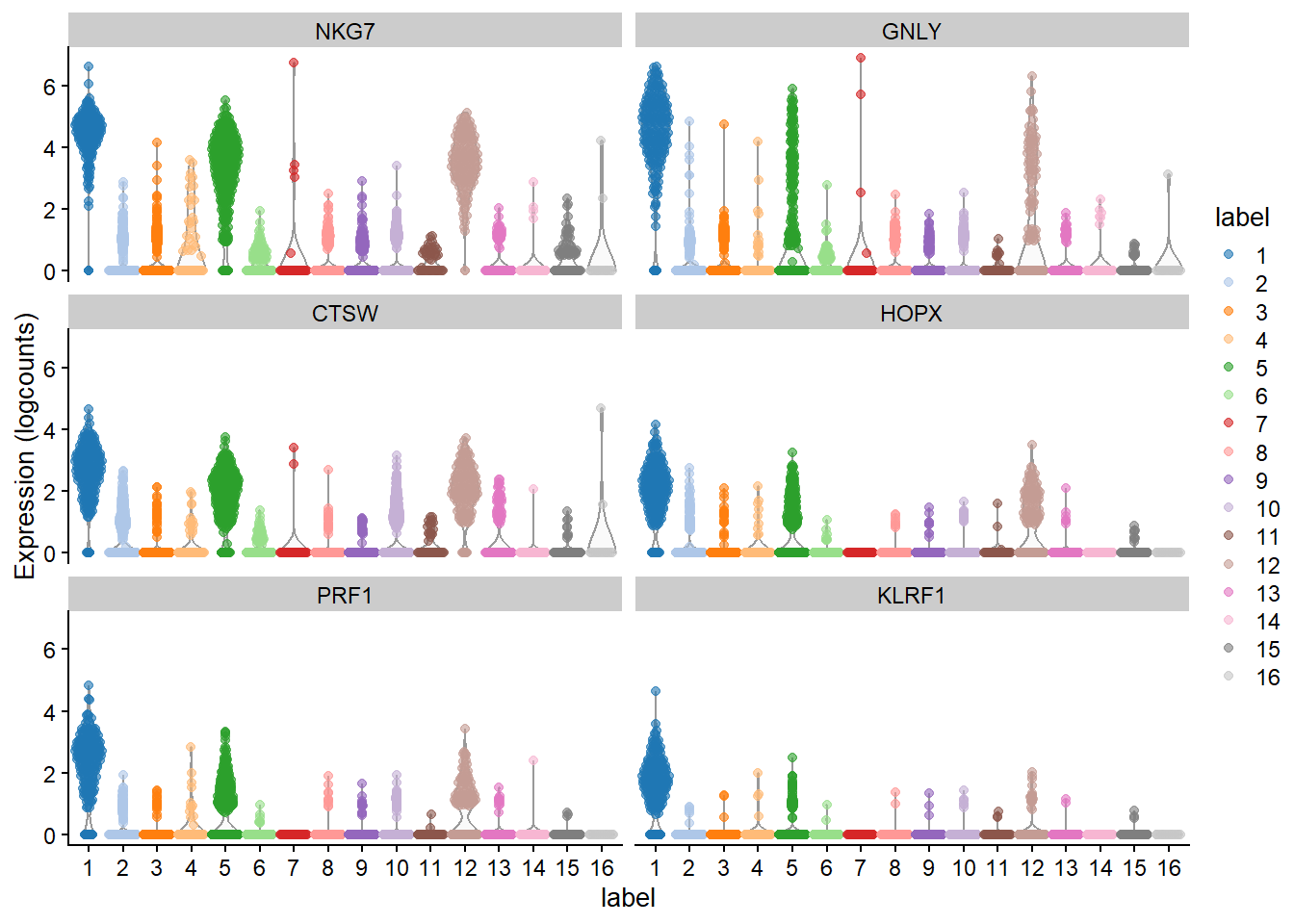
例如,根据**簇1**的`mean.AUC`作为排序
```{r}
chosen <- marker.info[["1"]]
ordered <- chosen[order(chosen$mean.AUC, decreasing=TRUE),]
head(ordered[,1:4]) # showing basic stats only, for brevity.
```
```{r fig-mean.AUC_cluster1}
library(scater)
plotExpression(sce.pbmc, features=head(rownames(ordered)),
x="label", colour_by="label")
```
## 效应值
Effect sizes for pairwise comparisons
### AUC
AUC 表示X簇中随机选择的观测值大于从其他簇中随机选择的观测值的概率。
值1对应于上调,其中X簇的所有值都大于其他簇的任何值;值0.5意味着分布的位置没有净差异;值0对应于下调。
AUC 与 Wilcoxon ranked sum test(又名 Mann-Whitney U 检验)中的U-statistic 密切相关。
```{r}
chosen
auc.only <- chosen[,grepl("AUC", colnames(chosen))]
auc.only[order(auc.only$mean.AUC,decreasing=TRUE),]
```
### LogFC.cohen
Cohen’s d是一种标准化的对数倍数变化(log-fold change),
正值表示该基因在簇中上调,负值表示下调,接近零的值表示差异不大。Cohen’s d大致类似于双样本t-检验的t-statistic
```{r}
cohen.only <- chosen[,grepl("logFC.cohen", colnames(chosen))]
cohen.only[order(cohen.only$mean.logFC.cohen,decreasing=TRUE),]
```
### logFC.detected
成对比较检测到表达值的细胞比例的对数倍数变化,这忽略了有关表达量的任何信息,只考虑是否检测到表达值。
正值表示与其他簇相比,X簇中表达基因的细胞比例更大。
```{r}
detect.only <- chosen[,grepl("logFC.detected", colnames(chosen))]
detect.only[order(detect.only$mean.logFC.detected,decreasing=TRUE),]
```
## summary
mean,与其他簇的平均值相比,对于簇X,较大的均值(>0 for the log-fold changes, >0.5 for the AUCs)表示在簇X中该基因上调。
median,与大多数 (>50%) 其他簇相比,较大的值表示该基因在X簇中上调。对于异常值, 中位数比均值提供更大的稳健性robustness,而均值可能是可取的,也可能是不可取的。 一方面,如果只有少数成对比较具有较大的效应,则中位数可以避免夸大效应值; 另一方面,它也会通过忽略少数具有相反效果的成对比较来夸大效应值。
minimum value,是鉴定上调基因最严格的。因为最小值是个大值表示与所有其他集群相比,该基因在X簇中上调。 相反,如果最小值很小(<0 for the log-fold changes, <0.5 for the AUCs),与至少一个其他簇相比,该基因在X簇中下调。
maximum value, 是识别上调基因最不严格的,因为与任何一个其他集群相比,如果在X簇中存在强烈上调,最大值可以是个大值。 相反,如果最大值很小,与所有其他集群相比,该基因在X簇中下调。
minimum rank,是所有成对比较中每个基因的最小排序。具体来说,根据效应值降序,在每个成对比较中对基因进行排序,然后为每个基因报告所有成对比较中的最小排序。如果一个基因的最小秩很小,在至少一个簇X与另一个簇的成对比较中,它是最上调的基因之一。
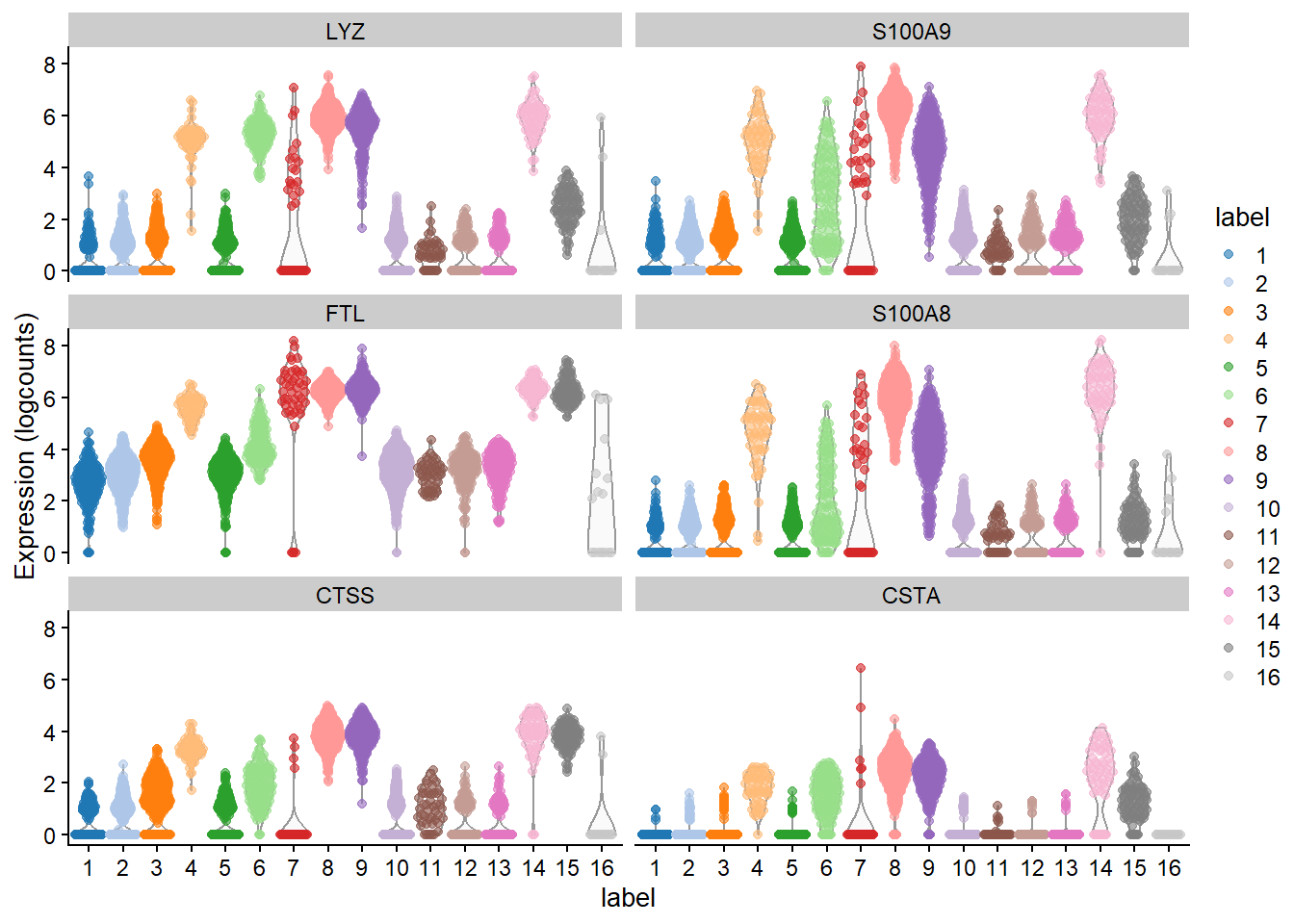
```{r}
chosen <- marker.info[["4"]] # using another cluster, for some variety.
ordered <- chosen[order(chosen$median.logFC.cohen,decreasing=TRUE),]
head(ordered[,1:4]) # showing basic stats only, for brevity.
```
```{r fig-median.logFC.cohen}
plotExpression(sce.pbmc, features=head(rownames(ordered)),
x="label", colour_by="label")
```
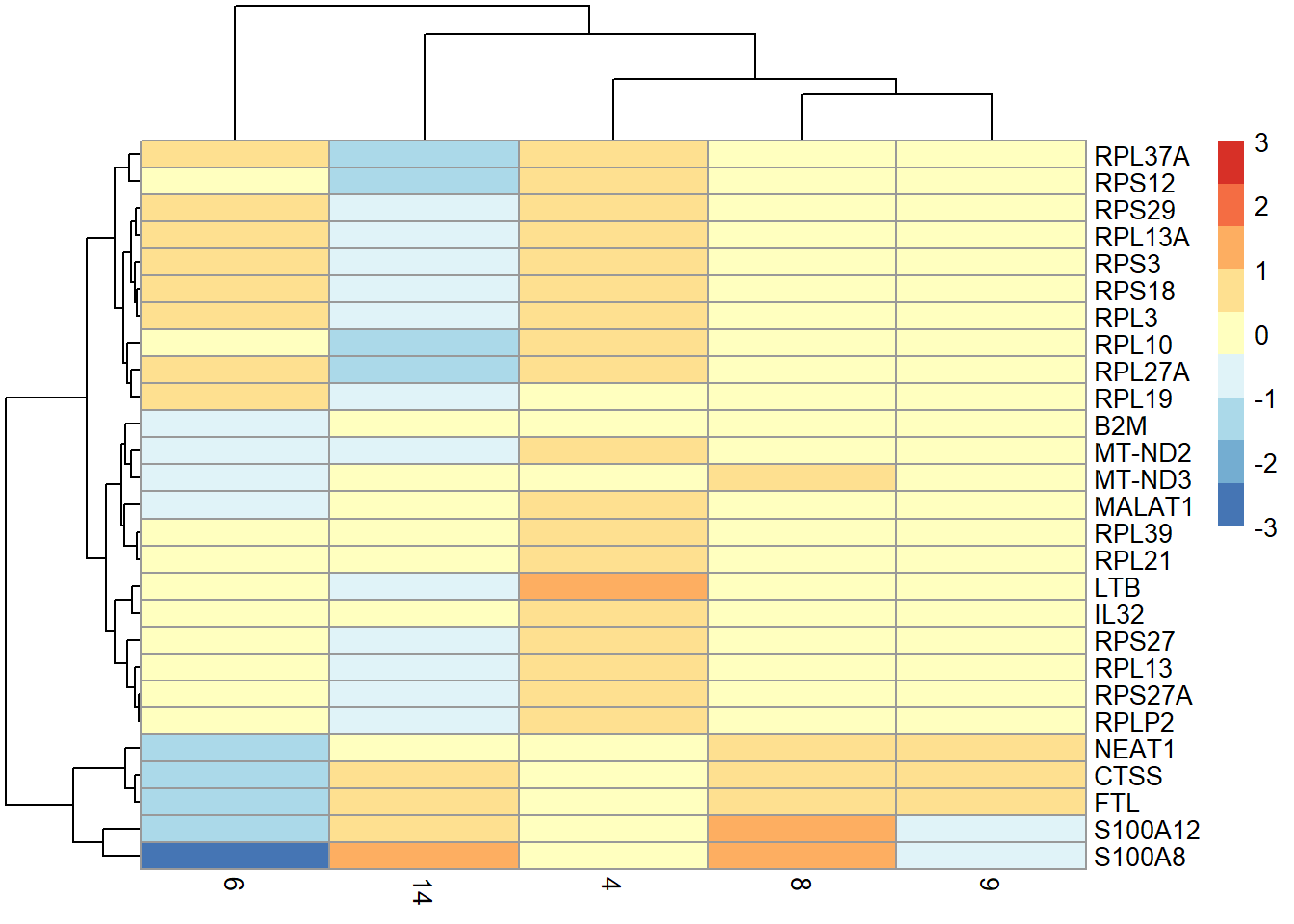
```{r fig-rank.logFC.cohen}
ordered <- chosen[order(chosen$rank.logFC.cohen),]
top.ranked <- ordered[ordered$rank.logFC.cohen <= 5,] # 最小排序小于或等于 5
top.ranked$rank.logFC.cohen
rownames(top.ranked)
plotGroupedHeatmap(sce.pbmc, features=rownames(top.ranked), group="label",
center=TRUE, zlim=c(-3, 3))
```
```{r}
# Omitting the decreasing=TRUE to focus on negative effects.
ordered <- chosen[order(chosen$median.logFC.cohen),1:4] #检查一些升序靠前的基因,看看与其他簇相比是否有任何一致的下调
head(ordered)
```
## full effects
设置 `full.stats=TRUE` 获取涉及特定聚类的所有成对比较的效应值
```{r}
# cluster 4 vs any other cluster
marker.info <- scoreMarkers(sce.pbmc, colLabels(sce.pbmc), full.stats=TRUE)
chosen <- marker.info[["4"]]
chosen$full.AUC
```
假设我们想确定将簇 4 与其他具有高 LYZ 表达的簇区分开来的基因。 对相关比较进行取子集,并对汇总统计量进行排序,以获得该子集中标记基因的排名。 这使我们能够轻松地表征密切相关的集群之间的细微差异。 为了说明这一点,与其他 高LYZ 簇相比,我们使用最小的排名`computeMinRank()`来识别簇 4 中排名靠前的 DE 基因。
```{r}
lyz.high <- c("4", "6", "8", "9", "14") # based on inspection of the previous Figure.
subset <- chosen$full.AUC[,colnames(chosen$full.AUC) %in% lyz.high]
to.show <- subset[computeMinRank(subset) <= 10,]
to.show
```
```{r fig-cluster4_vs_high-LYZ_DE}
plotGroupedHeatmap(sce.pbmc[,colLabels(sce.pbmc) %in% lyz.high],
features=rownames(to.show), group="label", center=TRUE, zlim=c(-3, 3))
```
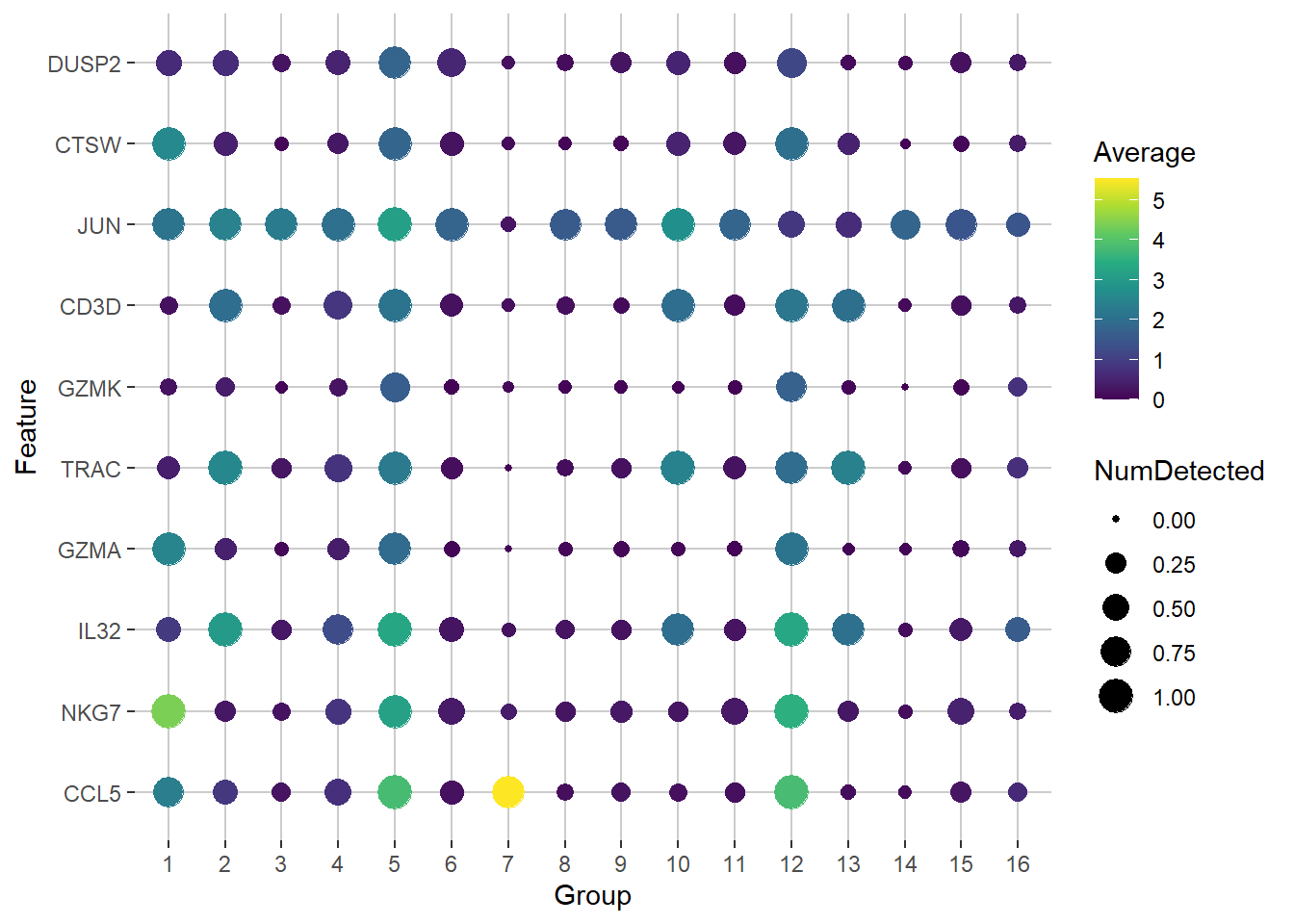
自定义汇总统计量例如,对于相对于其他集群的某个百分比(例如80%)上调的标记物感兴趣。
```{r}
stat <- MatrixGenerics::rowQuantiles(as.matrix(chosen$full.AUC), p=0.2)
chosen[order(stat, decreasing=TRUE), 1:4]
```
## log-fold change threshold
设置 `lfc= ` 来计算相对于对数倍数变化阈值的效应值
```{r}
marker.info.lfc <- scoreMarkers(sce.pbmc, colLabels(sce.pbmc), lfc=2)
chosen2 <- marker.info.lfc[["5"]]
chosen2 <- chosen2[order(chosen2$mean.AUC, decreasing=TRUE),]
chosen2[,c("self.average", "other.average", "mean.AUC")]
```
```{r}
plotDots(sce.pbmc, rownames(chosen2)[1:10], group="label")
```
## blocking factors
变异因素(例如,批次效应、性别差异)
` block= `
```{r loading-sce.416b_to_clustering}
#--- loading ---#
library(scRNAseq)
sce.416b <- LunSpikeInData(which="416b")
sce.416b$block <- factor(sce.416b$block)
#--- gene-annotation ---#
library(AnnotationHub)
ens.mm.v97 <- AnnotationHub()[["AH73905"]]
rowData(sce.416b)$ENSEMBL <- rownames(sce.416b)
rowData(sce.416b)$SYMBOL <- mapIds(ens.mm.v97, keys=rownames(sce.416b),
keytype="GENEID", column="SYMBOL")
rowData(sce.416b)$SEQNAME <- mapIds(ens.mm.v97, keys=rownames(sce.416b),
keytype="GENEID", column="SEQNAME")
library(scater)
rownames(sce.416b) <- uniquifyFeatureNames(rowData(sce.416b)$ENSEMBL,
rowData(sce.416b)$SYMBOL)
#--- quality-control ---#
mito <- which(rowData(sce.416b)$SEQNAME=="MT")
stats <- perCellQCMetrics(sce.416b, subsets=list(Mt=mito))
qc <- quickPerCellQC(stats, percent_subsets=c("subsets_Mt_percent",
"altexps_ERCC_percent"), batch=sce.416b$block)
sce.416b <- sce.416b[,!qc$discard]
#--- normalization ---#
library(scran)
sce.416b <- computeSumFactors(sce.416b)
sce.416b <- logNormCounts(sce.416b)
#--- variance-modelling ---#
dec.416b <- modelGeneVarWithSpikes(sce.416b, "ERCC", block=sce.416b$block)
chosen.hvgs <- getTopHVGs(dec.416b, prop=0.1)
#--- batch-correction ---#
library(limma)
assay(sce.416b, "corrected") <- removeBatchEffect(logcounts(sce.416b),
design=model.matrix(~sce.416b$phenotype), batch=sce.416b$block)
#--- dimensionality-reduction ---#
sce.416b <- runPCA(sce.416b, ncomponents=10, subset_row=chosen.hvgs,
exprs_values="corrected", BSPARAM=BiocSingular::ExactParam())
set.seed(1010)
sce.416b <- runTSNE(sce.416b, dimred="PCA", perplexity=10)
#--- clustering ---#
my.dist <- dist(reducedDim(sce.416b, "PCA"))
my.tree <- hclust(my.dist, method="ward.D2")
library(dynamicTreeCut)
my.clusters <- unname(cutreeDynamic(my.tree, distM=as.matrix(my.dist),
minClusterSize=10, verbose=0))
colLabels(sce.416b) <- factor(my.clusters)
```
```{r}
m.out <- scoreMarkers(sce.416b, colLabels(sce.416b), block=sce.416b$block)
```
在每个批次内进行成对比较,抵消了任何批次效应。然后使用加权平均数对各批次的效应值进行平均以获得每个成对比较的单个值,该加权平均数考虑了每个批次中参与成对比较的细胞数。对每个簇内外的平均对数表达和检测到的细胞比例进行了类似的校正。
```{r}
demo <- m.out[["1"]]
ordered <- demo[order(demo$median.logFC.cohen, decreasing=TRUE),]
ordered[,1:4]
```
```{r fig-block}
plotExpression(sce.416b, features=rownames(ordered)[1:6],
x="label", colour_by="block")
```
该参数`block=`适用于上面显示的所有效应值,并且对批次之间对数倍数变化或方差的差异具有鲁棒性。 但是,它假定每对簇至少存在于一个批次中。如果来自两个簇的细胞从未在同一批次中同时出现,则无法进行关联的成对比较,并且在计算汇总统计量时会忽略该比较。