4 SingleCellExperiment
4.1 父类SummarizeExperiment
SummarizeExperiment 如 Figure fig-SummarizeExperiment 所示
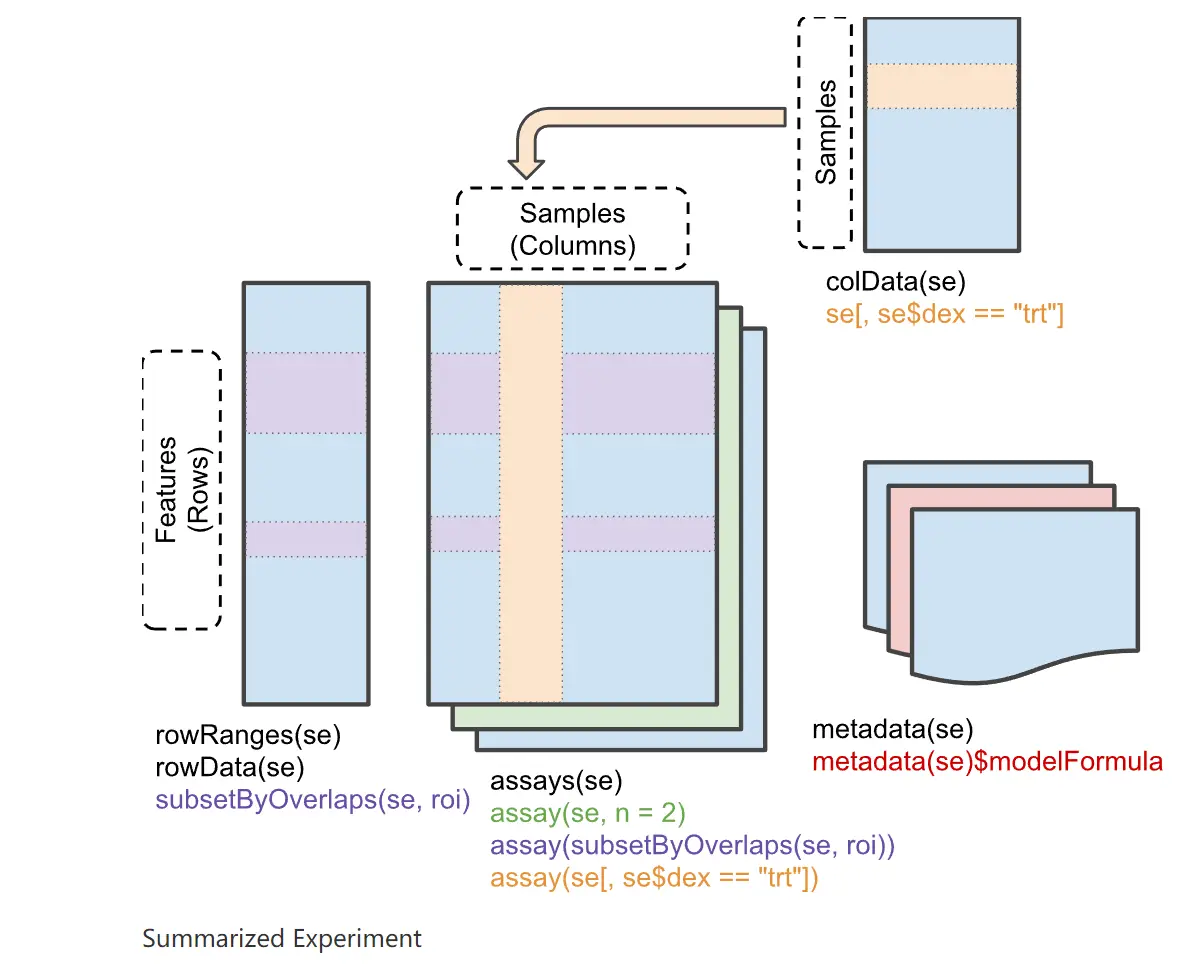
具体可参考 SummarizedExperiment docs 。
4.1.1 构造SummarizedExperiment实例
Code
# 计数矩阵
nrows <- 200
ncols <- 6
counts <- matrix(runif(nrows * ncols, 1, 1e4), nrows)
# 基因元数据
rowData <- GRanges(seqnames = rep(c("chr1", "chr2"), c(50, 150)),
ranges = IRanges(floor(runif(200, 1e5, 1e6)), width=100),
strand=sample(c("+", "-"), 200, TRUE),
feature_id=sprintf("ID%03d", 1:200))
rowData[1:6,]
#> GRanges object with 6 ranges and 1 metadata column:
#> seqnames ranges strand | feature_id
#> <Rle> <IRanges> <Rle> | <character>
#> [1] chr1 313525-313624 - | ID001
#> [2] chr1 431440-431539 - | ID002
#> [3] chr1 407554-407653 - | ID003
#> [4] chr1 669336-669435 - | ID004
#> [5] chr1 648855-648954 - | ID005
#> [6] chr1 209609-209708 + | ID006
#> -------
#> seqinfo: 2 sequences from an unspecified genome; no seqlengths
# 样本元数据
colData <- DataFrame(Treatment=rep(c("ChIP", "Input"), 3),
row.names=LETTERS[1:6])
colData
#> DataFrame with 6 rows and 1 column
#> Treatment
#> <character>
#> A ChIP
#> B Input
#> C ChIP
#> D Input
#> E ChIP
#> F Input
# 实验元数据
metadata <- "这是关于如何一个创建SE的说明对象"
se <- SummarizedExperiment(assays=list(counts=counts),
rowRanges=rowData,
colData=colData,
metadata=metadata)
se
#> class: RangedSummarizedExperiment
#> dim: 200 6
#> metadata(1): ''
#> assays(1): counts
#> rownames: NULL
#> rowData names(1): feature_id
#> colnames(6): A B ... E F
#> colData names(1): Treatment
4.2 类SingleCellExperiments
SingleCellExperiments如 Figure fig-sce 所示。
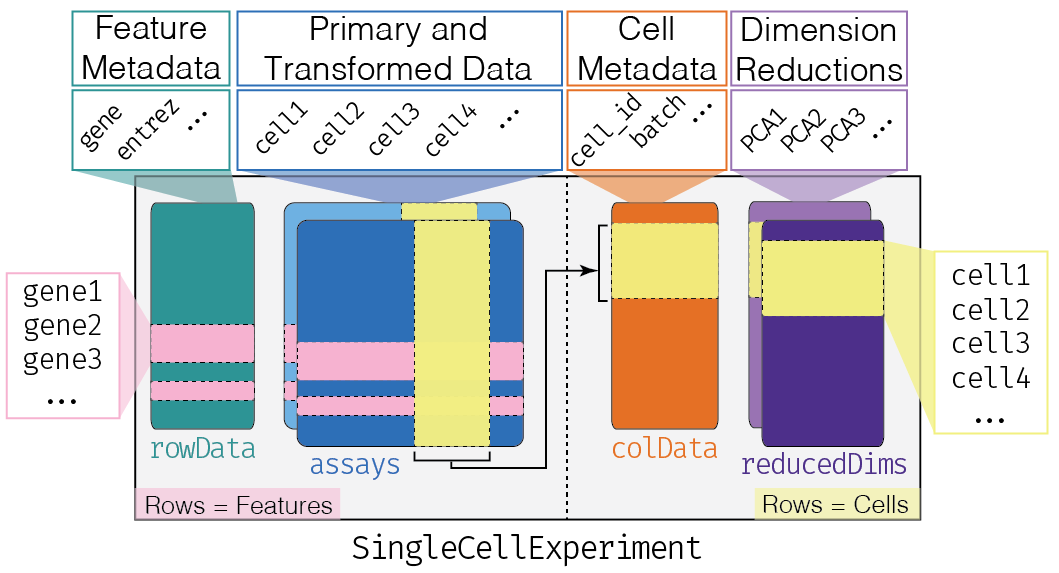
该数据结构实际上是从父类 SummarizedExperiment 继承。
Code
library(SingleCellExperiment)4.3 表达矩阵(raw/transformed counts)
要构造一个SingleCellExperiment对象,我们只需要导入assays ( Figure fig-sce ,蓝色框 ),其中行对应于特征(基因),列对应于样本(细胞)。下载:counts_Calero_20160113.tsv。
4.3.1 assays
Code
df <- read_tsv("data/OSCA/counts_Calero_20160113.tsv")
# 分离出spike-in RNA
spike.df <- df[str_detect(df$GeneID,"^ERCC-"),] #正则表达式,spike-in RNA
# 只考虑内源性基因
df<- df[str_detect(df$GeneID,"^ENSMUSG"),] #正则表达式,内源性RNA
# 分离基因长度
gene_length <- df$Length
df <- df |> column_to_rownames(var = "GeneID") # 行标识符
# 计数矩阵
mat<- as.matrix(df[,-1])
dim(mat)
#> [1] 46603 96ERCC = External RNA Controls Consortium ERCC就是一个专门为了定制一套spike-in RNA而成立的组织。
4.3.2 添加assays
然后使用函数 SingleCellExperiment() 以命名列表的形式提供数据,其中列表的每个对象都是一个矩阵。
Code
sce <- SingleCellExperiment(assays = list(counts = mat))
sce
#> class: SingleCellExperiment
#> dim: 46603 96
#> metadata(0):
#> assays(1): counts
#> rownames(46603): ENSMUSG00000102693 ENSMUSG00000064842 ...
#> ENSMUSG00000096730 ENSMUSG00000095742
#> rowData names(0):
#> colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
#> colData names(0):
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
4.3.2.1 自动添加assays
Code
sce <- scuttle::logNormCounts(sce) #对数变换标准化表达矩阵
sce # assays 多了一个 "logcounts"
#> class: SingleCellExperiment
#> dim: 46603 96
#> metadata(0):
#> assays(2): counts logcounts
#> rownames(46603): ENSMUSG00000102693 ENSMUSG00000064842 ...
#> ENSMUSG00000096730 ENSMUSG00000095742
#> rowData names(0):
#> colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
#> colData names(1): sizeFactor
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
dim(logcounts(sce))
#> [1] 46603 96
4.3.2.2 自定义添加assays
4.3.2.3 访问和切片
Code
#访问计数矩阵
assayNames(sce)
#> [1] "counts" "logcounts" "counts_100"
mat2 <- assay(sce, "counts") #通用
mat3 <- counts(sce) #特殊名称 counts
log_mat <- logcounts(sce)
assayNames(sce)
#> [1] "counts" "logcounts" "counts_100"
names(assays(sce))
#> [1] "counts" "logcounts" "counts_100"
# 切片
assays(sce) <- assays(sce)[1]
assayNames(sce)
#> [1] "counts"
4.4 cell metadata(colData)
为了进一步构造对象SingleCellExperiment,需要添加列元数据colData注释细胞或样本,该对象DataFrame中的行对应于细胞,列对应于样本元数据字段,例如原产地批次batch of origin、处理条件treatment condition等( Figure fig-sce ,橙色框)。下载:E-MTAB-5522.sdrf.txt(第2页)
4.4.1 colData
Code
coldata <- read_tsv("data/OSCA/E-MTAB-5522.sdrf.txt")
# 仅保留在计数矩阵 mat中的细胞 第44列=="counts_Calero_20160113.tsv"
coldata <-
coldata[coldata$`Derived Array Data File` == "counts_Calero_20160113.tsv", ]
# 仅保留部分列和设置行标识符
coldata <- DataFrame(
genotype=coldata$`Characteristics[genotype]`,
phenotype=coldata$`Characteristics[phenotype]`,
spike_in=coldata$`Factor Value[spike-in addition]`,
row.names = coldata$`Source Name`
)
coldata
#> DataFrame with 96 rows and 3 columns
#> genotype
#> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> ... ...
#> SLX-9555.N712_S505.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S506.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S507.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> phenotype spike_in
#> <character> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> ... ... ...
#> SLX-9555.N712_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S507.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 wild type phenotype Premixed添加之前确保colData的行名与计数矩阵的列名相同
4.4.2 添加colData
4.4.2.1 从头开始
Code
sce0 <- SingleCellExperiment(assays = list(counts=mat), colData=coldata)
sce0
colData(sce0)4.4.2.2 向现有对象添加
Code
sce
#> class: SingleCellExperiment
#> dim: 46603 96
#> metadata(0):
#> assays(1): counts
#> rownames(46603): ENSMUSG00000102693 ENSMUSG00000064842 ...
#> ENSMUSG00000096730 ENSMUSG00000095742
#> rowData names(0):
#> colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
#> colData names(1): sizeFactor
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
colData(sce) <- coldata
sce
#> class: SingleCellExperiment
#> dim: 46603 96
#> metadata(0):
#> assays(1): counts
#> rownames(46603): ENSMUSG00000102693 ENSMUSG00000064842 ...
#> ENSMUSG00000096730 ENSMUSG00000095742
#> rowData names(0):
#> colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
#> colData names(3): genotype phenotype spike_in
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):或者 分块添加
Code
sce1 <- SingleCellExperiment(list(counts=mat))
sce1$phenotype <- coldata$phenotype
colData(sce1)
#> DataFrame with 96 rows and 1 column
#> phenotype
#> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 wild type phenotype
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 wild type phenotype
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 wild type phenotype
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o..
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o..
#> ... ...
#> SLX-9555.N712_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o..
#> SLX-9555.N712_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o..
#> SLX-9555.N712_S507.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o..
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o..
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 wild type phenotype4.4.3 函数自动添加
某些函数在colData中返回额外样本元数据字段,自动添加列元数据。
Code
sce <- scuttle::addPerCellQC(sce) #quality control metrics质量控制指标
colData(sce)
#> DataFrame with 96 rows and 6 columns
#> genotype
#> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> ... ...
#> SLX-9555.N712_S505.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S506.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S507.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> phenotype spike_in
#> <character> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> ... ... ...
#> SLX-9555.N712_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S507.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 wild type phenotype Premixed
#> sum detected total
#> <numeric> <numeric> <numeric>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 854171 7617 854171
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 1044243 7520 1044243
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 1152450 8305 1152450
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 1193876 8142 1193876
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 1521472 7153 1521472
#> ... ... ... ...
#> SLX-9555.N712_S505.C89V9ANXX.s_1.r_1 203221 5608 203221
#> SLX-9555.N712_S506.C89V9ANXX.s_1.r_1 1059853 6948 1059853
#> SLX-9555.N712_S507.C89V9ANXX.s_1.r_1 1672343 6879 1672343
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1 1939537 7213 1939537
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 1436899 8469 1436899
sce
#> class: SingleCellExperiment
#> dim: 46603 96
#> metadata(0):
#> assays(1): counts
#> rownames(46603): ENSMUSG00000102693 ENSMUSG00000064842 ...
#> ENSMUSG00000096730 ENSMUSG00000095742
#> rowData names(0):
#> colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
#> colData names(6): genotype phenotype ... detected total
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
4.5 feature/gene metadata ( rowData)
添加行元数据rawData注释基因,DataFrame中每行对应一个基因,每列对应一个基因元数据字段,例如转录本长度、基因符号等注释。( Figure fig-sce ,绿色框)
4.5.0.1 rowData
Code
rowData(sce)
#> DataFrame with 46603 rows and 0 columns
rowData(sce)$Length <- gene_length
rowData(sce)
#> DataFrame with 46603 rows and 1 column
#> Length
#> <numeric>
#> ENSMUSG00000102693 1070
#> ENSMUSG00000064842 110
#> ENSMUSG00000051951 6094
#> ENSMUSG00000102851 480
#> ENSMUSG00000103377 2819
#> ... ...
#> ENSMUSG00000094431 100
#> ENSMUSG00000094621 121
#> ENSMUSG00000098647 99
#> ENSMUSG00000096730 3077
#> ENSMUSG00000095742 243某些函数在rowData中返回额外基因元数据字段,自动添加行元数据。
Code
sce <- scuttle::addPerFeatureQC(sce)
rowData(sce)
#> DataFrame with 46603 rows and 3 columns
#> Length mean detected
#> <numeric> <numeric> <numeric>
#> ENSMUSG00000102693 1070 0.0000000 0.00000
#> ENSMUSG00000064842 110 0.0000000 0.00000
#> ENSMUSG00000051951 6094 0.0000000 0.00000
#> ENSMUSG00000102851 480 0.0000000 0.00000
#> ENSMUSG00000103377 2819 0.0208333 1.04167
#> ... ... ... ...
#> ENSMUSG00000094431 100 0 0
#> ENSMUSG00000094621 121 0 0
#> ENSMUSG00000098647 99 0 0
#> ENSMUSG00000096730 3077 0 0
#> ENSMUSG00000095742 243 0 0
4.5.0.2 rowRanges
rowRanges:以GRangesList或 GRanges的形式保存基因组坐标,描述了特征(基因、基因组区域)的染色体、起始坐标和结束坐标。
Code
SummarizedExperiment::rowRanges(sce) #empty
#> GRangesList object of length 46603:
#> $ENSMUSG00000102693
#> GRanges object with 0 ranges and 0 metadata columns:
#> seqnames ranges strand
#> <Rle> <IRanges> <Rle>
#> -------
#> seqinfo: no sequences
#>
#> $ENSMUSG00000064842
#> GRanges object with 0 ranges and 0 metadata columns:
#> seqnames ranges strand
#> <Rle> <IRanges> <Rle>
#> -------
#> seqinfo: no sequences
#>
#> $ENSMUSG00000051951
#> GRanges object with 0 ranges and 0 metadata columns:
#> seqnames ranges strand
#> <Rle> <IRanges> <Rle>
#> -------
#> seqinfo: no sequences
#>
#> ...
#> <46600 more elements>填充 rowRanges的方式取决于在比对和定量过程中使用的生物体种类和注释文件。 常用的是Ensembl 标识符,因此我们可以使用rtracklayer从包含 Ensembl 注释的 GTF 文件中载入GRanges。下载:Mus_musculus.GRCm38.82.gtf.gz
Code
gene_data <- rtracklayer::import("data/OSCA/Mus_musculus.GRCm38.82.gtf.gz")
head(gene_data)
#> GRanges object with 6 ranges and 26 metadata columns:
#> seqnames ranges strand | source type score phase
#> <Rle> <IRanges> <Rle> | <factor> <factor> <numeric> <integer>
#> [1] 1 3073253-3074322 + | havana gene NA <NA>
#> [2] 1 3073253-3074322 + | havana transcript NA <NA>
#> [3] 1 3073253-3074322 + | havana exon NA <NA>
#> [4] 1 3102016-3102125 + | ensembl gene NA <NA>
#> [5] 1 3102016-3102125 + | ensembl transcript NA <NA>
#> [6] 1 3102016-3102125 + | ensembl exon NA <NA>
#> gene_id gene_version gene_name gene_source gene_biotype
#> <character> <character> <character> <character> <character>
#> [1] ENSMUSG00000102693 1 4933401J01Rik havana TEC
#> [2] ENSMUSG00000102693 1 4933401J01Rik havana TEC
#> [3] ENSMUSG00000102693 1 4933401J01Rik havana TEC
#> [4] ENSMUSG00000064842 1 Gm26206 ensembl snRNA
#> [5] ENSMUSG00000064842 1 Gm26206 ensembl snRNA
#> [6] ENSMUSG00000064842 1 Gm26206 ensembl snRNA
#> havana_gene havana_gene_version transcript_id
#> <character> <character> <character>
#> [1] OTTMUSG00000049935 1 <NA>
#> [2] OTTMUSG00000049935 1 ENSMUST00000193812
#> [3] OTTMUSG00000049935 1 ENSMUST00000193812
#> [4] <NA> <NA> <NA>
#> [5] <NA> <NA> ENSMUST00000082908
#> [6] <NA> <NA> ENSMUST00000082908
#> transcript_version transcript_name transcript_source transcript_biotype
#> <character> <character> <character> <character>
#> [1] <NA> <NA> <NA> <NA>
#> [2] 1 4933401J01Rik-001 havana TEC
#> [3] 1 4933401J01Rik-001 havana TEC
#> [4] <NA> <NA> <NA> <NA>
#> [5] 1 Gm26206-201 ensembl snRNA
#> [6] 1 Gm26206-201 ensembl snRNA
#> havana_transcript havana_transcript_version tag
#> <character> <character> <character>
#> [1] <NA> <NA> <NA>
#> [2] OTTMUST00000127109 1 basic
#> [3] OTTMUST00000127109 1 basic
#> [4] <NA> <NA> <NA>
#> [5] <NA> <NA> basic
#> [6] <NA> <NA> basic
#> transcript_support_level exon_number exon_id exon_version
#> <character> <character> <character> <character>
#> [1] <NA> <NA> <NA> <NA>
#> [2] NA <NA> <NA> <NA>
#> [3] NA 1 ENSMUSE00001343744 1
#> [4] <NA> <NA> <NA> <NA>
#> [5] NA <NA> <NA> <NA>
#> [6] NA 1 ENSMUSE00000522066 1
#> ccds_id protein_id protein_version
#> <character> <character> <character>
#> [1] <NA> <NA> <NA>
#> [2] <NA> <NA> <NA>
#> [3] <NA> <NA> <NA>
#> [4] <NA> <NA> <NA>
#> [5] <NA> <NA> <NA>
#> [6] <NA> <NA> <NA>
#> -------
#> seqinfo: 45 sequences from an unspecified genome; no seqlengths
# 整理数据
gene_data <- gene_data[gene_data$type=="gene"]
names(gene_data) <- gene_data$gene_id
#DataFrame:mcols(gene_data)
is.gene.related <- str_detect(colnames(mcols(gene_data)),"gene_") # 6 TRUE
mcols(gene_data) <- mcols(gene_data)[,is.gene.related]
mcols(gene_data) # 46603 × 6
#> DataFrame with 46603 rows and 6 columns
#> gene_id gene_version gene_name
#> <character> <character> <character>
#> ENSMUSG00000102693 ENSMUSG00000102693 1 4933401J01Rik
#> ENSMUSG00000064842 ENSMUSG00000064842 1 Gm26206
#> ENSMUSG00000051951 ENSMUSG00000051951 5 Xkr4
#> ENSMUSG00000102851 ENSMUSG00000102851 1 Gm18956
#> ENSMUSG00000103377 ENSMUSG00000103377 1 Gm37180
#> ... ... ... ...
#> ENSMUSG00000094431 ENSMUSG00000094431 1 CAAA01205117.1
#> ENSMUSG00000094621 ENSMUSG00000094621 1 CAAA01098150.1
#> ENSMUSG00000098647 ENSMUSG00000098647 1 CAAA01064564.1
#> ENSMUSG00000096730 ENSMUSG00000096730 6 Vmn2r122
#> ENSMUSG00000095742 ENSMUSG00000095742 1 CAAA01147332.1
#> gene_source gene_biotype havana_gene_version
#> <character> <character> <character>
#> ENSMUSG00000102693 havana TEC 1
#> ENSMUSG00000064842 ensembl snRNA NA
#> ENSMUSG00000051951 ensembl_havana protein_coding 2
#> ENSMUSG00000102851 havana processed_pseudogene 1
#> ENSMUSG00000103377 havana TEC 1
#> ... ... ... ...
#> ENSMUSG00000094431 ensembl miRNA NA
#> ENSMUSG00000094621 ensembl miRNA NA
#> ENSMUSG00000098647 ensembl miRNA NA
#> ENSMUSG00000096730 ensembl protein_coding NA
#> ENSMUSG00000095742 ensembl protein_coding NA
#rownames(sce) 46603行 观测基因
SummarizedExperiment::rowRanges(sce) <- gene_data[rownames(sce)]
SummarizedExperiment::rowRanges(sce)[1:6,]
#> GRanges object with 6 ranges and 6 metadata columns:
#> seqnames ranges strand | gene_id
#> <Rle> <IRanges> <Rle> | <character>
#> ENSMUSG00000102693 1 3073253-3074322 + | ENSMUSG00000102693
#> ENSMUSG00000064842 1 3102016-3102125 + | ENSMUSG00000064842
#> ENSMUSG00000051951 1 3205901-3671498 - | ENSMUSG00000051951
#> ENSMUSG00000102851 1 3252757-3253236 + | ENSMUSG00000102851
#> ENSMUSG00000103377 1 3365731-3368549 - | ENSMUSG00000103377
#> ENSMUSG00000104017 1 3375556-3377788 - | ENSMUSG00000104017
#> gene_version gene_name gene_source
#> <character> <character> <character>
#> ENSMUSG00000102693 1 4933401J01Rik havana
#> ENSMUSG00000064842 1 Gm26206 ensembl
#> ENSMUSG00000051951 5 Xkr4 ensembl_havana
#> ENSMUSG00000102851 1 Gm18956 havana
#> ENSMUSG00000103377 1 Gm37180 havana
#> ENSMUSG00000104017 1 Gm37363 havana
#> gene_biotype havana_gene_version
#> <character> <character>
#> ENSMUSG00000102693 TEC 1
#> ENSMUSG00000064842 snRNA <NA>
#> ENSMUSG00000051951 protein_coding 2
#> ENSMUSG00000102851 processed_pseudogene 1
#> ENSMUSG00000103377 TEC 1
#> ENSMUSG00000104017 TEC 1
#> -------
#> seqinfo: 45 sequences from an unspecified genome; no seqlengths
sce
#> class: SingleCellExperiment
#> dim: 46603 96
#> metadata(0):
#> assays(1): counts
#> rownames(46603): ENSMUSG00000102693 ENSMUSG00000064842 ...
#> ENSMUSG00000096730 ENSMUSG00000095742
#> rowData names(6): gene_id gene_version ... gene_biotype
#> havana_gene_version
#> colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
#> colData names(6): genotype phenotype ... detected total
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):4.5.1 其他元数据
其他注释以命名列表存储在metadata 中。 例如,实验元数据,高度可变基因(highly variable genes)。。。,缺点是与表达矩阵的行或列的操作不同步。
Code
DEG_up <- c("gene_x", "gene_y")
metadata(sce) <- list(HVGs = DEG_up)
metadata(sce)
#> $HVGs
#> [1] "gene_x" "gene_y"
DEG_down <- c("gene_a", "gene_b")
metadata(sce)$DEG_down <- DEG_down
metadata(sce)
#> $HVGs
#> [1] "gene_x" "gene_y"
#>
#> $DEG_down
#> [1] "gene_a" "gene_b"
sce
#> class: SingleCellExperiment
#> dim: 46603 96
#> metadata(2): HVGs DEG_down
#> assays(1): counts
#> rownames(46603): ENSMUSG00000102693 ENSMUSG00000064842 ...
#> ENSMUSG00000096730 ENSMUSG00000095742
#> rowData names(6): gene_id gene_version ... gene_biotype
#> havana_gene_version
#> colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
#> colData names(6): genotype phenotype ... detected total
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):4.6 子集和组合
4.6.1 子集
Code
first.10 <- sce[,1:10]
ncol(counts(first.10)) # 计数矩阵仅有 10 列
#> [1] 10
colData(first.10) # only 10 rows.
#> DataFrame with 10 rows and 6 columns
#> genotype
#> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S507.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S508.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S517.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N702_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N702_S503.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> phenotype spike_in
#> <character> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> SLX-9555.N701_S507.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> SLX-9555.N701_S508.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> SLX-9555.N701_S517.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N702_S502.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N702_S503.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> sum detected total
#> <numeric> <numeric> <numeric>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 854171 7617 854171
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 1044243 7520 1044243
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 1152450 8305 1152450
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 1193876 8142 1193876
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 1521472 7153 1521472
#> SLX-9555.N701_S507.C89V9ANXX.s_1.r_1 866705 6828 866705
#> SLX-9555.N701_S508.C89V9ANXX.s_1.r_1 608581 6966 608581
#> SLX-9555.N701_S517.C89V9ANXX.s_1.r_1 1113526 8634 1113526
#> SLX-9555.N702_S502.C89V9ANXX.s_1.r_1 1308250 8364 1308250
#> SLX-9555.N702_S503.C89V9ANXX.s_1.r_1 778605 8665 778605只想要野生型细胞
Code
wt.only <- sce[, sce$phenotype == "wild type phenotype"]
ncol(counts(wt.only))
#> [1] 48
colData(wt.only)
#> DataFrame with 48 rows and 6 columns
#> genotype phenotype
#> <character> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> SLX-9555.N701_S517.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> SLX-9555.N702_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> ... ... ...
#> SLX-9555.N711_S517.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> SLX-9555.N712_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> SLX-9555.N712_S503.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> SLX-9555.N712_S504.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype
#> spike_in sum detected total
#> <character> <numeric> <numeric> <numeric>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 ERCC+SIRV 854171 7617 854171
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ERCC+SIRV 1044243 7520 1044243
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 ERCC+SIRV 1152450 8305 1152450
#> SLX-9555.N701_S517.C89V9ANXX.s_1.r_1 ERCC+SIRV 1113526 8634 1113526
#> SLX-9555.N702_S502.C89V9ANXX.s_1.r_1 ERCC+SIRV 1308250 8364 1308250
#> ... ... ... ... ...
#> SLX-9555.N711_S517.C89V9ANXX.s_1.r_1 Premixed 1317671 8581 1317671
#> SLX-9555.N712_S502.C89V9ANXX.s_1.r_1 Premixed 1736189 9687 1736189
#> SLX-9555.N712_S503.C89V9ANXX.s_1.r_1 Premixed 1521132 8983 1521132
#> SLX-9555.N712_S504.C89V9ANXX.s_1.r_1 Premixed 1759166 8480 1759166
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 Premixed 1436899 8469 1436899只想保留蛋白质编码基因
Code
coding.only <- sce[rowData(sce)$gene_biotype == "protein_coding",]
nrow(counts(coding.only))
#> [1] 22013
rowData(coding.only)
#> DataFrame with 22013 rows and 6 columns
#> gene_id gene_version gene_name
#> <character> <character> <character>
#> ENSMUSG00000051951 ENSMUSG00000051951 5 Xkr4
#> ENSMUSG00000025900 ENSMUSG00000025900 9 Rp1
#> ENSMUSG00000025902 ENSMUSG00000025902 12 Sox17
#> ENSMUSG00000033845 ENSMUSG00000033845 12 Mrpl15
#> ENSMUSG00000025903 ENSMUSG00000025903 13 Lypla1
#> ... ... ... ...
#> ENSMUSG00000079808 ENSMUSG00000079808 3 AC168977.1
#> ENSMUSG00000095041 ENSMUSG00000095041 6 PISD
#> ENSMUSG00000063897 ENSMUSG00000063897 3 DHRSX
#> ENSMUSG00000096730 ENSMUSG00000096730 6 Vmn2r122
#> ENSMUSG00000095742 ENSMUSG00000095742 1 CAAA01147332.1
#> gene_source gene_biotype havana_gene_version
#> <character> <character> <character>
#> ENSMUSG00000051951 ensembl_havana protein_coding 2
#> ENSMUSG00000025900 ensembl_havana protein_coding 2
#> ENSMUSG00000025902 ensembl_havana protein_coding 6
#> ENSMUSG00000033845 ensembl_havana protein_coding 3
#> ENSMUSG00000025903 ensembl_havana protein_coding 3
#> ... ... ... ...
#> ENSMUSG00000079808 ensembl protein_coding NA
#> ENSMUSG00000095041 ensembl protein_coding NA
#> ENSMUSG00000063897 ensembl protein_coding NA
#> ENSMUSG00000096730 ensembl protein_coding NA
#> ENSMUSG00000095742 ensembl protein_coding NA4.6.2 组合
按列组合,假设所有涉及的对象都具有相同的行注释值和兼容的列注释字段
Code
sce2 <- cbind(sce, sce)
ncol(counts(sce2)) # twice as many columns
#> [1] 192
colData(sce2) # twice as many rows
#> DataFrame with 192 rows and 6 columns
#> genotype
#> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> ... ...
#> SLX-9555.N712_S505.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S506.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S507.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
#> phenotype spike_in
#> <character> <character>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 wild type phenotype ERCC+SIRV
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. ERCC+SIRV
#> ... ... ...
#> SLX-9555.N712_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S507.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. Premixed
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 wild type phenotype Premixed
#> sum detected total
#> <numeric> <numeric> <numeric>
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 854171 7617 854171
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 1044243 7520 1044243
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 1152450 8305 1152450
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 1193876 8142 1193876
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 1521472 7153 1521472
#> ... ... ... ...
#> SLX-9555.N712_S505.C89V9ANXX.s_1.r_1 203221 5608 203221
#> SLX-9555.N712_S506.C89V9ANXX.s_1.r_1 1059853 6948 1059853
#> SLX-9555.N712_S507.C89V9ANXX.s_1.r_1 1672343 6879 1672343
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1 1939537 7213 1939537
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1 1436899 8469 1436899按行组合,假设所有对象都具有相同的列注释值和兼容的行注释字段。
Code
sce2 <- rbind(sce, sce)
nrow(counts(sce2)) # twice as many rows
#> [1] 93206
rowData(sce2) # twice as many rows
#> DataFrame with 93206 rows and 6 columns
#> gene_id gene_version gene_name
#> <character> <character> <character>
#> ENSMUSG00000102693 ENSMUSG00000102693 1 4933401J01Rik
#> ENSMUSG00000064842 ENSMUSG00000064842 1 Gm26206
#> ENSMUSG00000051951 ENSMUSG00000051951 5 Xkr4
#> ENSMUSG00000102851 ENSMUSG00000102851 1 Gm18956
#> ENSMUSG00000103377 ENSMUSG00000103377 1 Gm37180
#> ... ... ... ...
#> ENSMUSG00000094431 ENSMUSG00000094431 1 CAAA01205117.1
#> ENSMUSG00000094621 ENSMUSG00000094621 1 CAAA01098150.1
#> ENSMUSG00000098647 ENSMUSG00000098647 1 CAAA01064564.1
#> ENSMUSG00000096730 ENSMUSG00000096730 6 Vmn2r122
#> ENSMUSG00000095742 ENSMUSG00000095742 1 CAAA01147332.1
#> gene_source gene_biotype havana_gene_version
#> <character> <character> <character>
#> ENSMUSG00000102693 havana TEC 1
#> ENSMUSG00000064842 ensembl snRNA NA
#> ENSMUSG00000051951 ensembl_havana protein_coding 2
#> ENSMUSG00000102851 havana processed_pseudogene 1
#> ENSMUSG00000103377 havana TEC 1
#> ... ... ... ...
#> ENSMUSG00000094431 ensembl miRNA NA
#> ENSMUSG00000094621 ensembl miRNA NA
#> ENSMUSG00000098647 ensembl miRNA NA
#> ENSMUSG00000096730 ensembl protein_coding NA
#> ENSMUSG00000095742 ensembl protein_coding NA4.7 单细胞特定字段
4.7.1 降维 reducedDims
降维结果保存在一个列表中,列表的每一个对象是一个代表计数矩阵的低维的数值矩阵,其中行表示计数矩阵的列(如细胞),列表示维度。
PCA
sce
#> class: SingleCellExperiment
#> dim: 46603 96
#> metadata(2): HVGs DEG_down
#> assays(1): counts
#> rownames(46603): ENSMUSG00000102693 ENSMUSG00000064842 ...
#> ENSMUSG00000096730 ENSMUSG00000095742
#> rowData names(6): gene_id gene_version ... gene_biotype
#> havana_gene_version
#> colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
#> colData names(6): genotype phenotype ... detected total
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
sce <- scater::logNormCounts(sce)
sce <- scater::runPCA(sce)
dim(reducedDim(sce, "PCA"))
#> [1] 96 50tSNE
sce <- scater::runTSNE(sce, perplexity = 0.1)
#> Perplexity should be lower than K!
head(reducedDim(sce, "TSNE"))
#> TSNE1 TSNE2
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 548.4412 -219.68769
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 642.7446 33.43924
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 898.8680 151.30741
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 -1191.2253 -135.97727
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 -827.7419 -958.72969
#> SLX-9555.N701_S507.C89V9ANXX.s_1.r_1 -294.8303 -867.20137UMAP
sce <- scater::runUMAP(sce)
head(reducedDim(sce,"UMAP"))
#> UMAP1 UMAP2
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 -2.915830 -4.855348
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 -2.255113 -4.339309
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 -2.745740 -5.377293
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 -3.738179 -7.343237
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 4.381729 7.615599
#> SLX-9555.N701_S507.C89V9ANXX.s_1.r_1 4.752352 7.368036手动添加UMAP
u <- uwot::umap(t(logcounts(sce)), n_neighbors = 2)
reducedDim(sce, "UMAP_uwot") <- u
reducedDims(sce) # Now stored in the object.
#> List of length 4
#> names(4): PCA TSNE UMAP UMAP_uwot
head(reducedDim(sce, "UMAP_uwot"))
#> [,1] [,2]
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 2.528333 -1.22023178
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 1.831814 0.91103887
#> SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 3.373020 0.06124734
#> SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 3.981762 -2.07711912
#> SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 -7.307404 -5.91599203
#> SLX-9555.N701_S507.C89V9ANXX.s_1.r_1 -5.876931 -4.32073761reduceDims()
reducedDims(sce)
#> List of length 4
#> names(4): PCA TSNE UMAP UMAP_uwot4.7.2 替代试验 Alternative Experiments
SingleCellExperiment提供了”替代实验”的概念,其是一组不同特征但同一组样本/细胞的数据。经典应用是存储加标转录(spike-in transcripts)的每细胞计数,能够保留这些数据以供下游使用,但要将其与保存的内源性基因计数分离,因为此类替代特征通常需要单独处理。
Code
spike.df <- spike.df |> column_to_rownames("GeneID")
spike_length <- spike.df$Length
spike.mat<- as.matrix(spike.df[,-1])
spike.mat[1:2,1:2]
#> SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> ERCC-00002 12948
#> ERCC-00003 220
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
#> ERCC-00002 11287
#> ERCC-00003 911首先创建一个单独的对象SummarizedExperiment
Code
spike_se <- SummarizedExperiment(list(counts=spike.mat))
spike_se
#> class: SummarizedExperiment
#> dim: 92 96
#> metadata(0):
#> assays(1): counts
#> rownames(92): ERCC-00002 ERCC-00003 ... ERCC-00170 ERCC-00171
#> rowData names(0):
#> colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
#> SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
#> SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
#> colData names(0):替代实验概念确保单细胞数据集的所有相关方面都可以保存在单个对象中,并且确保我们的加标数据与内源性基因的数据同步。
Code
sub <- sce[,1:2] # retain only two samples.
altExp(sub, "spike")
#> class: SummarizedExperiment
#> dim: 92 2
#> metadata(0):
#> assays(1): counts
#> rownames(92): ERCC-00002 ERCC-00003 ... ERCC-00170 ERCC-00171
#> rowData names(0):
#> colnames(2): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
#> SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
#> colData names(0):任何SummarizedExperiment对象都可以存储为alternative Experiment, 包括另一个 SingleCellExperiment。
4.7.3 缩放因子sizeFactors
Code
# 反卷积deconvolution-based size factors
sce <- scran::computeSumFactors(sce)
summary(sizeFactors(sce))
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 0.1513 0.7417 0.9478 1.0000 1.1239 3.5583手动添加
Code
# library size-derived factors
sizeFactors(sce) <- scater::librarySizeFactors(sce)
summary(sizeFactors(sce))
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 0.1703 0.7657 0.9513 1.0000 1.1064 3.60504.7.4 列标签
该函数允许我们获取或设置每个细胞标签的向量或因子,通常对应于由无监督聚类分析的分组 或从分类算法预测细胞类型身份。
Code
colLabels(sce) <- scran::clusterCells(sce, use.dimred="PCA")
table(colLabels(sce))
#>
#> 1 2
#> 47 49