The library size is defined as the total sum of counts across all relevant features for each cell. Cells with small library sizes are of low quality. 文库大小定义为每个细胞所有基因的计数总和。库小表示细胞低质量。
The number of expressed features in each cell is defined as the number of endogenous genes with non-zero counts for that cell. Any cell with very few expressed genes is likely to be of poor quality. 每个细胞中表达的特征数被定义为该细胞具有非零计数的内源性基因的数量。低表达特征数表示细胞低质量。
The proportion of reads mapped to spike-in transcripts is calculated relative to the total count across all features (including spike-ins) for each cell.High proportions are indicative of poor-quality cells。 相对于每个细胞所有特征(包括spike-in)的总计数,计算映射到spike-in transcripts中的读数比例。高比例表示细胞质量差
In the absence of spike-in transcripts, the proportion of reads mapped to genes in the mitochondrial genome can be used. High proportions are indicative of poor-quality cells。在没有spike-in transcripts的情况下,可以使用映射到线粒体基因组中的读数比例。高比例表示细胞质量差。
标记低质量的细胞,并将它们保留在下游分析中,但是保留会降低方差建模的准确性,例如,需要使用更多的 PC 来抵消早期 PC 是由低质量细胞和其他细胞之间的差异引起的事实。
Code
marked<-sce.416bmarked$discard<-reasons$discard
Source Code
# 质量控制scRNA-seq数据中的低质量细胞文库(library)来源,如解离过程中的细胞损伤或cDNA文库制备失败(如低效的逆转录或PCR扩增),这些通常表现为总计数低、表达基因少、线粒体或尖峰比例高的"细胞"。这些低质量的细胞文库会导致下游分析出现误导性结果。为了提高生物学意义,需要在分析开始时删除低质量的细胞(cell),此步骤称为细胞的质量控制 (QC)。一个关键假设是QC指标与每个细胞的生物状态无关。 较差的值(如低文库大小、高线粒体比例)被认为是由技术因素而不是生物过程引起的,这意味着低质量细胞过滤不会在下游分析中歪曲生物学意义。严重违反这一假设可能会导致细胞类型的丧失。```{r include=FALSE}conflicts_prefer(GenomicRanges::setdiff)conflicts_prefer(dplyr::filter)``````{r loading-sce.416b}library(scRNAseq)library(ensembldb)sce.416b <-LunSpikeInData(which="416b") sce.416b$block <- factor(sce.416b$block)sce.416b```## QC metrics- **The library size** is defined as the total sum of counts across all relevant features for each cell. Cells with small library sizes are of low quality. 文库大小定义为每个细胞所有基因的计数总和。库小表示细胞低质量。- **The number of expressed features in each cell** is defined as the number of endogenous genes with non-zero counts for that cell. Any cell with very few expressed genes is likely to be of poor quality. 每个细胞中表达的特征数被定义为该细胞具有非零计数的内源性基因的数量。低表达特征数表示细胞低质量。- **The proportion of reads mapped to spike-in transcripts** is calculated relative to the total count across all features (including spike-ins) for each cell.High proportions are indicative of poor-quality cells。 相对于每个细胞所有特征(包括**spike-in**)的总计数,计算映射到**spike-in transcripts**中的读数比例。高比例表示细胞质量差- **In the absence of spike-in transcripts, the proportion of reads mapped to genes in the mitochondrial genome** can be used. High proportions are indicative of poor-quality cells。在没有**spike-in transcripts**的情况下,可以使用映射到线粒体基因组中的读数比例。高比例表示细胞质量差。函数`perCellQCMetrics()`可以计算以上 QC 指标,其中`sum`列包含每个细胞文库大小的总计数,`detected`列包含检测到的基因数。 `subsets_Mito_percent`列包含映射到线粒体基因组的读取比例,`altexps_ERCC_percent`列包含映射到 ERCC 转录本的读取比例。```{r}location <- SummarizedExperiment::rowRanges(sce.416b)locationis.mito <-any(seqnames(location)=="MT")library(scuttle)df <-perCellQCMetrics(sce.416b, subsets=list(Mito=is.mito))colnames(df)summary(df$sum)summary(df$subsets_Mito_percent)summary(df$altexps_ERCC_percent)```或者 `addPerCellQC()`计算每个细胞的 QC 统计数据并将其附加到`SingleCellExperiment`的`colData` ,将所有相关信息保留在单个对象`SingleCellExperiment`中以供以后操作。```{r}sce.416b <-addPerCellQCMetrics(sce.416b, subsets=list(Mito=is.mito))dim(colData(sce.416b))colnames(colData(sce.416b))sce.416b```## 识别低质量细胞### 固定阈值识别低质量细胞的最简单方法是对 QC 指标应用固定阈值。 例如,如果细胞的文库大小低于 100,000 次读取;表达少于5,000个基因;spike-in比例超过10%;或线粒体比例高于10%,我们可能会认为它们的质量较低。```{r}qc.lib <- df$sum <1e5qc.nexprs <- df$detected <5e3qc.spike <- df$altexps_ERCC_percent >10qc.mito <- df$subsets_Mito_percent >10discard <- qc.lib | qc.nexprs | qc.spike | qc.mito# 汇总tibble(LibSize=sum(qc.lib), NExprs=sum(qc.nexprs),SpikeProp=sum(qc.spike), MitoProp=sum(qc.mito), Total=sum(discard))```### 自适应阈值假设大部分数据集由高质量的细胞组成,然后,我们根据所有细胞中每个指标的绝对中位差(median absolute deviation ,MAD)来识别各种QC指标的异常值细胞。默认情况下,如果一个值距离中位数超过3个MAD,可以将其视为异常值。即此类过滤器将保留 99% 遵循正态分布的非异常值。```{r}#?perCellQCFiltersreasons <-perCellQCFilters(df, sub.fields=c("subsets_Mito_percent","altexps_ERCC_percent"))colSums(as.matrix(reasons)) # apply(as.matrix(reasons),2,sum)summary(reasons$discard)# 提取自适应阈值attr(reasons$low_lib_size, "thresholds")attr(reasons$low_n_features, "thresholds")```### outlyingness根据每个细胞的 QC 指标识别高维空间中的异常值`高outlyingness`低质量, 在一定程度上降低了可解释性```{r}stats <-cbind(log10(df$sum), log10(df$detected), df$subsets_Mito_percent, df$altexps_ERCC_percent)library(robustbase)outlying <-adjOutlyingness(stats, only.outlyingness =TRUE)multi.outlier <-isOutlier(outlying, type ="higher")summary(multi.outlier)```## 诊断图观察QC指标的分布( @fig-diagnostic-plots )以识别低质量细胞是一种很好的做法。 在最理想的情况下,我们会看到正态分布,这些分布可以证明异常值检测中使用的 3 MAD 阈值是合理的。在非正态分布中的很大一部分细胞表明QC指标可能与某些生物状态相关,可能导致过滤过程中不同细胞类型的损失;或者与细胞亚群的文库制备不一致。```{r}#| label: fig-diagnostic-plots#| fig-cap: "数据集中每个批次和表型的 QC 指标分布。每个点代表一个细胞,并分别根据其是否被丢弃而着色。"colData(sce.416b) <-cbind(colData(sce.416b), df)sce.416b$block <-factor(sce.416b$block)sce.416b$phenotype <-ifelse(grepl("induced", sce.416b$phenotype),"induced", "wild type")sce.416b$discard <- reasons$discardlibrary(scater)gridExtra::grid.arrange(plotColData(sce.416b, x="block", y="sum", colour_by="discard",other_fields="phenotype") +facet_wrap(~phenotype) +scale_y_log10() +ggtitle("Total count"),plotColData(sce.416b, x="block", y="detected", colour_by="discard", other_fields="phenotype") +facet_wrap(~phenotype) +scale_y_log10() +ggtitle("Detected features"),plotColData(sce.416b, x="block", y="subsets_Mito_percent", colour_by="discard", other_fields="phenotype") +facet_wrap(~phenotype) +ggtitle("Mito percent"),plotColData(sce.416b, x="block", y="altexps_ERCC_percent", colour_by="discard", other_fields="phenotype") +facet_wrap(~phenotype) +ggtitle("ERCC percent"),ncol=1)```另一种有用的诊断图涉及线粒体计数与其他 QC 指标的比例。 目的是确认没有细胞同时具有大量总计数和大量线粒体计数,以确保不会无意中去除恰好具有高度代谢活性的高质量细胞(例如肝细胞)。```{r}#| label: loading-sce.zeisel #| code-summary: "loading ZeiselBrainData"sce.zeisel <- scRNAseq::ZeiselBrainData()sce.zeisellibrary(scater)sce.zeisel <-aggregateAcrossFeatures(sce.zeisel, id=sub("_loc[0-9]+$", "", rownames(sce.zeisel)))#--- gene-annotation ---#library(org.Mm.eg.db)rowData(sce.zeisel)$Ensembl <-mapIds(org.Mm.eg.db, keys=rownames(sce.zeisel), keytype="SYMBOL", column="ENSEMBL")``````{r}#| label: fig-mito_pct/sum#| fig-cap: "ZeiselBrainData中映射线粒体转录本的 UMI 百分比与 UMI 总数作图。每个点代表一个细胞,并根据它是否被视为低质量并丢弃分组。"sce.zeisel <-addPerCellQC( sce.zeisel,subsets=list(Mt=rowData(sce.zeisel)$featureType=="mito"))qc <-quickPerCellQC(colData(sce.zeisel), sub.fields=c("altexps_ERCC_percent", "subsets_Mt_percent"))sce.zeisel$discard <- qc$discardplotColData(sce.zeisel, x="sum", y="subsets_Mt_percent", colour_by="discard")``````{r}#| label: fig-mito_pct/ERCC#| fig-cap: "ZeiselBrainData中映射线粒体转录本的 UMI 比例与映射到 spike-in transcripts 的 UMI 比例作图。每个点代表一个细胞,并根据它是否被视为低质量并丢弃分组。"plotColData(sce.zeisel, x="altexps_ERCC_percent", y="subsets_Mt_percent",colour_by="discard")```## 细胞过滤对于常规分析,删除是最直接的方法,避免保留低质量细胞的弊端。```{r}filtered <- sce.416b[,!reasons$discard]```标记低质量的细胞,并将它们保留在下游分析中,但是保留会降低方差建模的准确性,例如,需要使用更多的 PC 来抵消早期 PC 是由低质量细胞和其他细胞之间的差异引起的事实。```{r}marked <- sce.416bmarked$discard <- reasons$discard```
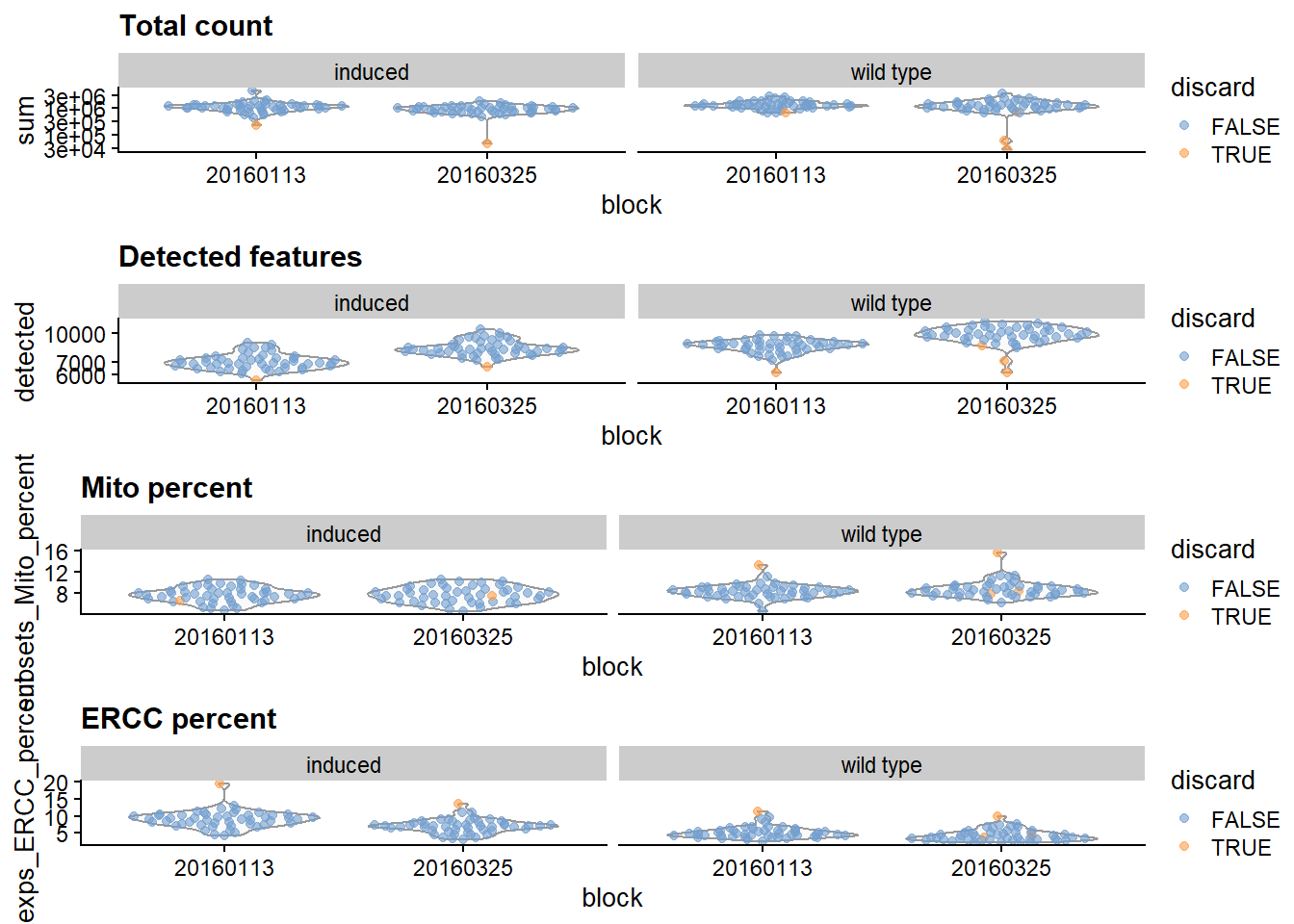
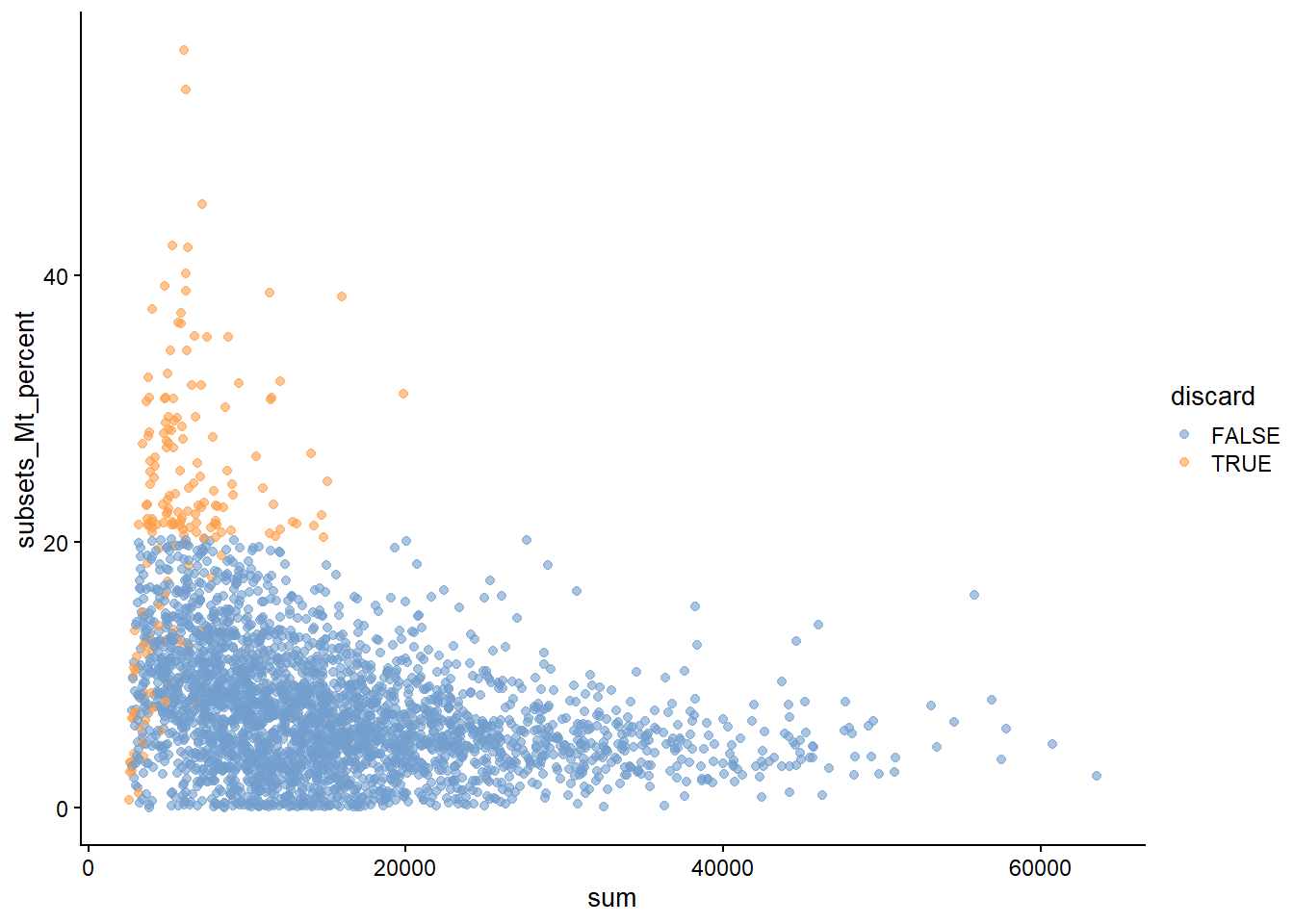 {#fig-mito_pct/sum width=672}
{#fig-mito_pct/sum width=672}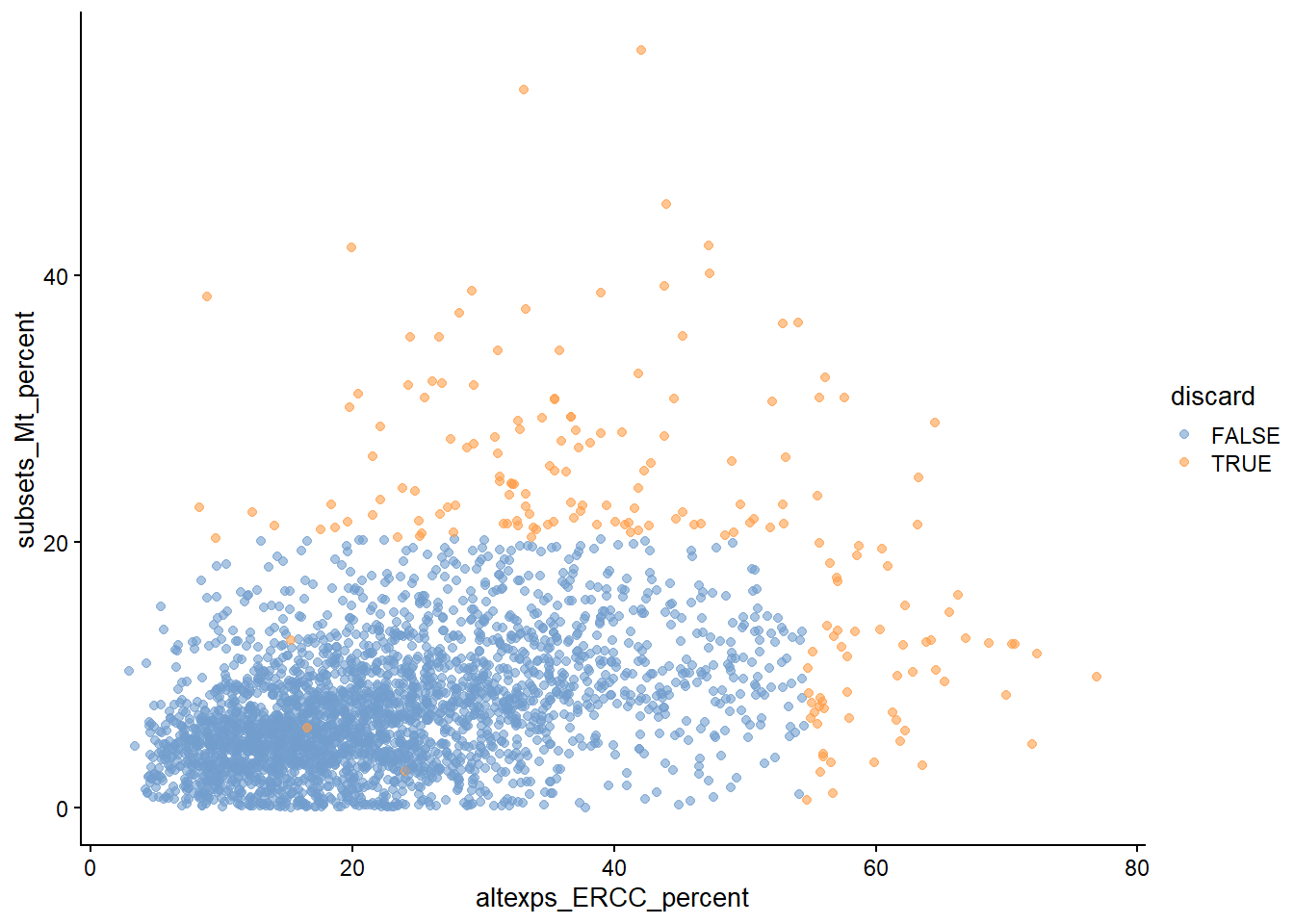 {#fig-mito_pct/ERCC width=672}
{#fig-mito_pct/ERCC width=672}